another good review of the drugs
Best Circulatory Review (Chest 2002;121:877)
Best review of Guyton Graph and adding kidney to the mix
Interpretation of Blood Pressure
There is no descending limb on a starling curve

Best article on VR and CVP physiology (Anesthes 2008;108(4):735)
Shock Dx: Feel the Feet Look at the Neck Veins Echo ABD Uts/Fast C-XR Fingerstick 12 Lead Rx:NorEpi Epi Phenylephrine Bolus Ephedrine Bolus CaCl Fluid Dobutamine Decadron Monitoring:
Preload=LVEDV
there is no descending limb of starling, fluid can’t affect hemodynamics
but you can cause tissue and lung edema which can effect tissue ox
preload doesn’t = fluid responsiveness
venoconstriction can release 60-80% of the blood volume, increasing preload Afterload-impedance to ventricular ejection Lusitropism-the ability to relax during diastole afterload dominates in the failing heart to determine CO Norepinephine leaches fluids out of the vesselsIncreasing MAP with norepi beyond 65 does not increase microcirc flow (Crit Care 2009;13:R92)

Bathmotropic is one of five adjectives used to describe various qualities of the cardiac cycle; the other four are: inotropic chronotropic dromotropic and lusiotropic. In an article in the American Journal of Medical Sciences these five terms were described as the five fundamental properties of the heart.[3] While Bathmotropic, as used herein has been defined as pertaining to modification of the excitability of the heart it can equally well refer to modification of the irritability of heart muscle, and the two terms are frequently used interchangeably.[4]
Pressors improve Cardiac Output
Norepi improves CI, SVI, and LVSWI in patients unresponsive to dobutamine, but doesn’t cause improvement in patients not on dobutamine b/c they already have a high cardiac index (Martin Crit Care Med 1999;27(9):1708)
Starling Curves
Best Review (Inten Care Med 2009;35:45)
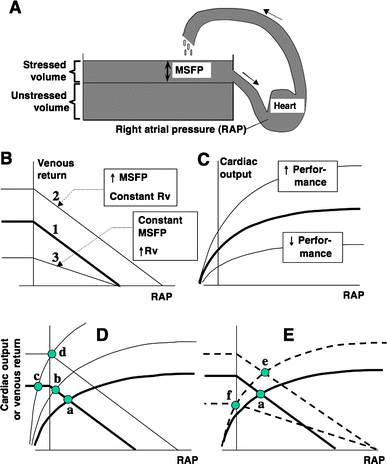
Fig. 2 Interactions of venous return and cardiac function. a Magders representation of the circulatory system. Modified from [8], with permission. MSFP mean systemic filling pressure. Detailed explanations in the text (beginning of Sect. Venous return curve). b Venous return curves (later part of Sect. Venous return curve). c cardiac function curves (Sect. Cardiac function curve). d Guytons graphical analysis of cardiac output regulation (Sect. Graphical analysis of cardiac output/venous return). e Potential effects of generalized venoconstriction on cardiac output (last paragraph of Sect. Graphical analysis of cardiac output/venous return). In panels be, RAP designates right atrial pressure relative to atmosphere
Circulatory Model The care of the critically ill hemodynamically unstable patientoften proceeds along the following two parallel paths: physiologic resuscitation and differential diagnosis investigation. Frequently, the initial physiologic characterization and the subsequent physiologic response to therapy contribute to establishing the definitive diagnosis and initiating optimal treatment. Accordingly, the utilization of a universally applicable physiologic modelof the circulation that allows for the expeditious applicationof resuscitative and diagnostic strategies is beneficial. Thisis particularly pertinent to MPE, given the acknowledged difficultyin deciphering the process, the potential for rapid lethality,and controversies in treatment. A fundamental understandingand review of basic hemodynamic principles is imperative toappreciate the pathophysiologic alterations induced by variousdisease states. Utilizing Poiseuilles law, conventionalhemodynamics conceptualize the circulatory system as an opencylindrical conduit with cardiac output (CO) defined as a functionof pressure gradients (mean arterial pressure [MAP] – rightatrial pressure [RAP]) against resistance (Fig 2). However,recognizing that CO is pulsatile, it is useful to devise a modelthat includes a hydraulic pump.
Poiseuilles law representing the relationships among flow (Qflow), pressure, and resistance.
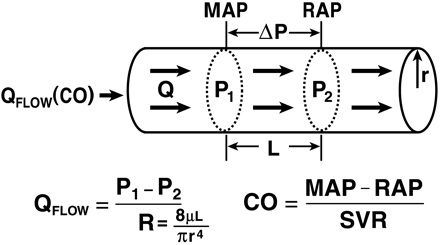 Figure 3 illustrates a three-compartment circulatory modelthat conceptualizes the circulatory system as a hydraulic pump composed of a right heart pump linked in series to a left heartpump. As a consequence of this serial hydraulic alignment, COcannot exceed venous return (VR) and vice versa. In other words,left heart output cannot exceed right heart output, which allowsfor the conceptualization of both pumps as a single hydraulicunit. The hydraulic pump is primed with volume from the venouscapacitance bed [ie, the volume reservoir] and empties intothe arterial impedance bed (ie, the resistive element). Guytonet al49 recognized that the pressure gradient for VR is theratio of pressure in the venous capacitance bed (PVC) to theRAP (VR = PVC – RAP), thus establishing the integral role ofthe right atrium (RA) as a coupler of the venous system andcardiac hydraulic circulation. The graphic solution of thisobservation is depicted in Figure 3 . PVC is a function of venousvolume and vascular tone, which must exceed the RAP to maintainVR. The RAP provides not only an assessment of the pressurein the right heart but an indirect gauge of the pressure inthe venous capacitance system. Thus, the circulatory systemcan be defined as a three-compartment model; a capacitance bedthat provides volume to a hydraulic pump that generates flowinto an impedance bed. Any hemodynamic abnormality can be characterizedby disturbances of one or more of these three variables. Thesurrogates for venous capacitance pressure, hydraulic pump function,and impedance are RAP, CO, and systemic vascular resistance(SVR), respectively. Invasive monitoring is frequently not inplace on initial presentation, and, given the controversiessurrounding its risks and benefits,50 it is prudent to utilizereadily available physical examination surrogates to definethe model variables. Estimation of the RAP from the internaljugular vein approximates the pressure in the venous capacitancesystem, and the pulse character and temperature of the extremitiesapproximate impedance (resistance). Warm flushed extremitieswith a wide pulse pressure indicate low impedance (ie, resistance),whereas cool constricted extremities with a narrow thready pulsesuggest high impedance (ie, resistance). The latter is a consequenceof the catecholamine-mediated vasoconstriction that is initiatedto create perfusion pressure gradients to redistribute and optimizethe low-flow state. In shock patients, flow and resistance arealmost uniformly reciprocal (Qflow x resistance = pressure orCO x SVR = BP). Therefore, the initial assessment of impedance(ie, resistance) allows for the inferential derivation of hydraulicflow (ie, CO). Obviously, invasive monitoring will be neededif the physical examination findings cannot be well-characterized.Representative examples are illustrated in Figure 3 .
Figure 3 illustrates a three-compartment circulatory modelthat conceptualizes the circulatory system as a hydraulic pump composed of a right heart pump linked in series to a left heartpump. As a consequence of this serial hydraulic alignment, COcannot exceed venous return (VR) and vice versa. In other words,left heart output cannot exceed right heart output, which allowsfor the conceptualization of both pumps as a single hydraulicunit. The hydraulic pump is primed with volume from the venouscapacitance bed [ie, the volume reservoir] and empties intothe arterial impedance bed (ie, the resistive element). Guytonet al49 recognized that the pressure gradient for VR is theratio of pressure in the venous capacitance bed (PVC) to theRAP (VR = PVC – RAP), thus establishing the integral role ofthe right atrium (RA) as a coupler of the venous system andcardiac hydraulic circulation. The graphic solution of thisobservation is depicted in Figure 3 . PVC is a function of venousvolume and vascular tone, which must exceed the RAP to maintainVR. The RAP provides not only an assessment of the pressurein the right heart but an indirect gauge of the pressure inthe venous capacitance system. Thus, the circulatory systemcan be defined as a three-compartment model; a capacitance bedthat provides volume to a hydraulic pump that generates flowinto an impedance bed. Any hemodynamic abnormality can be characterizedby disturbances of one or more of these three variables. Thesurrogates for venous capacitance pressure, hydraulic pump function,and impedance are RAP, CO, and systemic vascular resistance(SVR), respectively. Invasive monitoring is frequently not inplace on initial presentation, and, given the controversiessurrounding its risks and benefits,50 it is prudent to utilizereadily available physical examination surrogates to definethe model variables. Estimation of the RAP from the internaljugular vein approximates the pressure in the venous capacitancesystem, and the pulse character and temperature of the extremitiesapproximate impedance (resistance). Warm flushed extremitieswith a wide pulse pressure indicate low impedance (ie, resistance),whereas cool constricted extremities with a narrow thready pulsesuggest high impedance (ie, resistance). The latter is a consequenceof the catecholamine-mediated vasoconstriction that is initiatedto create perfusion pressure gradients to redistribute and optimizethe low-flow state. In shock patients, flow and resistance arealmost uniformly reciprocal (Qflow x resistance = pressure orCO x SVR = BP). Therefore, the initial assessment of impedance(ie, resistance) allows for the inferential derivation of hydraulicflow (ie, CO). Obviously, invasive monitoring will be neededif the physical examination findings cannot be well-characterized.Representative examples are illustrated in Figure 3 .

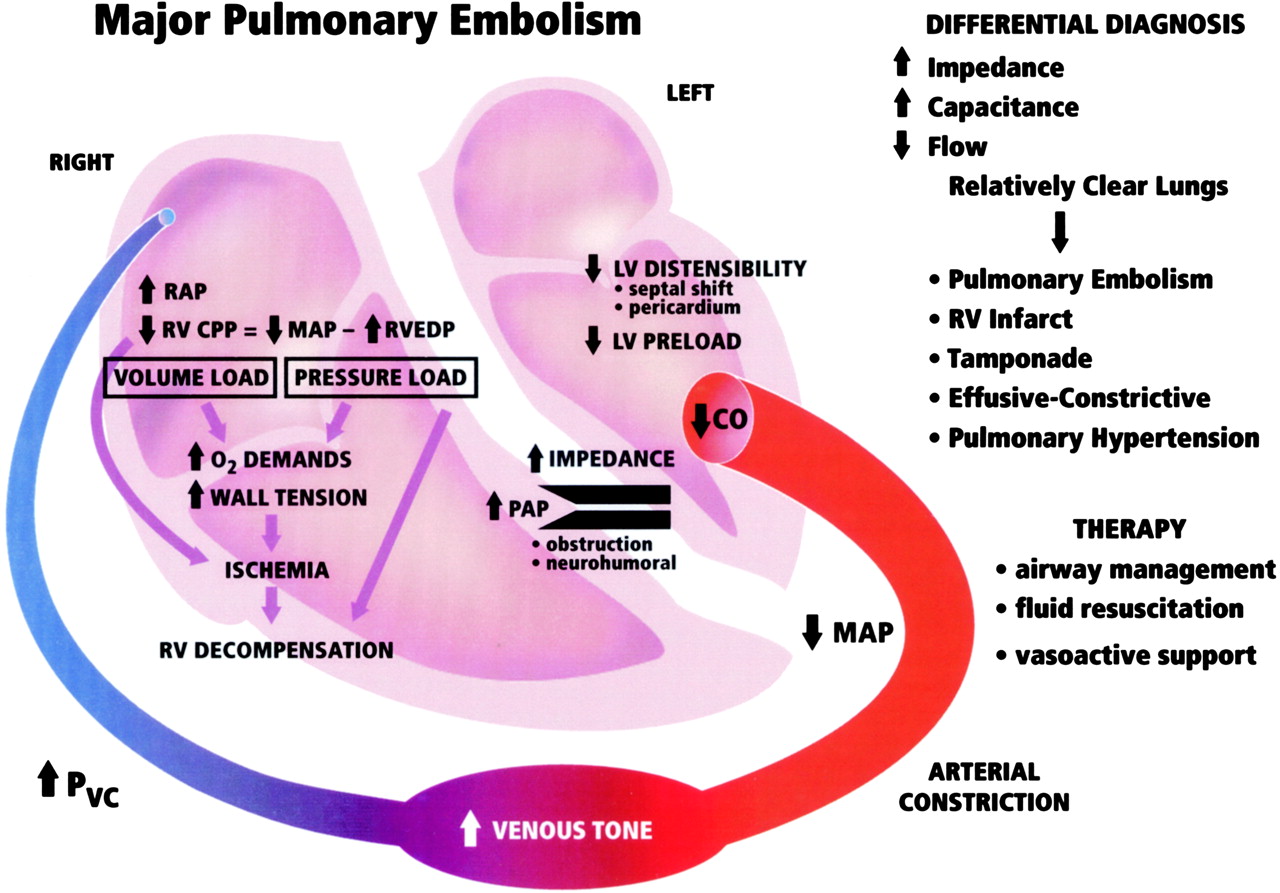
Pathophysiology
Mechanism of Cardiac Failure Cardiac failure from MPE results from a combination of the increased wall stress and cardiac ischemia that comprise RV function andimpair left ventricular (LV) output. Research from animal modelsand evidence from clinical investigations clearly demonstratethat the impact of embolic material on the pulmonary vascularoutflow tract precipitates an increase in RV impedance. Thisinitiates the vicious pathophysiologic cycle depicted in Figure 4. The degree of increase in RV impedance is predominantlyrelated to the interaction of the mechanical obstruction withthe underlying cardiopulmonary status.515253 Additional factorsreported to contribute to increased RV impedance include pulmonary vasoconstriction induced by neural reflexes,54 the releaseof humoral factors55 from platelets (ie, serotonin and plateletactivating factor), plasma (ie, thrombin and vasoactive peptidesC3a, C5a), tissue (ie, histamine), and systemic arterial hypoxia.56 The acute development of this increased RV impedance constitutesa pressure afterload on the RV and has multiple effects on RVand LV function.
Given the reciprocal relationship between RV stroke volume and vascular load, RV stroke volume will diminish with increasingload.57 Initially, the compensatory maintenance of CO is achievedby a combination of catecholamine-driven tachycardia and theutilization of the Frank-Starling preload reserve (the latterbeing responsible for RV dilatation). This increase in RV cavitarypressure and radius serves to significantly increase RV wallstress (wall stress = pressure x radius). This is the primarydeterminant of RV oxygen uptake, thus creating the potential for RV ischemia. With increasing RV load and wall stress, RVsystolic function becomes depressed and CO begins to decrease.Interestingly, systemic BP may be adequately maintained by systemicvasoconstriction at this point.58 From the point of initialCO depression, it has been reported59 that increases in loadsufficient to further decrease CO by 20% will result in a disproportionateincrease in end-systolic volume compared to end-diastolic volume.Afterload mismatch has been used to describe the phenomenonof RV pressure work exceeding RV volume work in this setting.60 As a consequence of this mismatch, LV preload will decrease,given the ventricular alignment in series. LV preload is additionallyimpaired by decreased LV distensibility as a consequence ofa leftward shift of the interventricular septum and of pericardialrestraint, both of which are related to the degree of RV dilatation.616263 It also has been suggested that MPE may impair LV function independentlyof preload mechanisms.64 In the presence of declining LV forwardflow, MAP can be maintained only by catecholamine-induced vasoconstriction.A further decrease in LV flow results in systemic hypotension.RV coronary perfusion pressure (CPP) depends on the gradientbetween the MAP and the RV subendocardial pressure. Decreasesin MAP associated with increases in RV end-diastolic pressure(RVEDP) impair the subendocardial perfusion and oxygen supply.Elevated right-sided pressures can further impair coronary perfusionand LV distensibility by increasing coronary venous pressure.65 Increased oxygen demands associated with elevated wall stresscoupled with decreased oxygen supply have been shown to precipitateRV ischemia, which is thought to be the cause of RV failure.66 Clinical evidence of RV infarction as a consequence of the precedingcondition has been demonstrated in patients with and withoutobstructive coronary disease.676869 A reversal of PE-inducedRV ischemia and RV failure can be accomplished by the infusionof vasoconstrictors to raise aortic pressure and to increasethe coronary perfusion gradient.6670
Translation of the pathophysiology of MPE into the previously discussed three-compartment hydraulic model of the circulationis shown in Figure 5 . Catecholamine-induced venoconstrictionincreases the PVC to maintain a pressure gradient for VR in response to the PE-induced RAP elevation. The impairment ofRV hydraulic pump function compromises LV hydraulic output,which is manifested as systemic arterial hypotension. Thus,the model variables would reveal an increased RAP, a decreasedCO, and an increased SVR. The clinical correlates would be jugularvenous distention, a thready pulse, and cool extremities, respectively.
1st Question: Is this patient in shock; is the shock adequately resuscitated
2nd Question: Does the patient need fluid
Markers of regional perfusion are really the answer to the first question
Neligan Notes
CV system consists of a pump (cardiogenic), tubing (distributive), and fluid (hypovolemic)
Shock is malfunction of at least one of the above. It is hypotension with signs of end organ failure
Another way to think about it is a failure of one of the following: stroke volume, heart, or peripheral vascular resistance and a failure of compensation of the other two.

Dysoxia-when the production of ATP is limited by oxygen supply
Low Stroke Volume
Inadequate Venous Return
Signs= a lingering tachycardia, cold peripheries or a pulse oximeter that is not reading, oliguria, low CVP, a large base excess on blood gas analysis, a lactic acidosis. In this state, patient can become hypotensive from medications such as sedatives.
Diastolic Dysfunction = stiff heart, requiring higher filling pressure to achieve normal volume. 2) Diastolic Dysfunction: loss of left ventricular compliance impairs its ability to receive blood. This disorder most commonly results from systolic dysfunction, and as a consequence of myocardial fibrosis for example due to ischemia or hypertension. Diastolic dysfunction is characterized by the requirement of higher filling pressures to achieve normal filling volumes, while the heart is less compliant and receptive to blood. Aggressive volume loading of patients with diastolic dysfunction frequently results in backward heart failure, causing acute pulmonary edema. Cardiac inflow obstruction is caused by a pericardial (tamponade) or intrathoracic process (PEEP), or a lesion within the heart itself (mitral stenosis). 3) Cardiac inflow obstruction: occurs either due to a constriction around the heart, a pericardial or intrathoracic process, or a lesion within the heart itself. Pericardial injuries include pericardial effusion or hematoma constrictive pericarditis an acute crisis associated with a pericardial injury is called tamponade. Tamponade is diagnosed as a tetrad of shock, clear lung fields, inaudible or muffled heart sounds, and an increase in the jugular venous pulse waveform on inspiration. An often forgotten but extremely common cause of hypotension is excessive intrathoracic pressure. This can be transmitted from within the alveolar space as with positive end expiratory pressure (PEEP) and gas trapping in airway obstruction (auto-PEEP), or within the pleural space Pneumothorax, hemothorax or, if the patient is in extremis, tension Pneumothorax. Intracardiac lesions may also cause inflow obstruction; these include mitral and tricuspid stenosis or thrombosis, and atrial myxoma.
Systolic dysfunction is pump failure from ischemia or overload
Cardiac outflow obstruction is caused by pulmonary embolism, aortic stenosis, aortic crossclamps 2. Outflow obstruction: there are two major sites that cardiac outflow may be blocked: at the level of the aortic valve (aortic stenosis) or within the low pressure (at thus easily occluded) pulmonary circulation pulmonary embolism. The former can be diagnosed on the basis of history, ECG and classic murmur. The latter may be more difficult to diagnose. Useful information includes risk (cancer, immobility, deep venous thrombosis, lack of prophylaxis, pelvic and hip surgery), ECG changes (right sided RVH, sinus tachycardia, atrial fibrillation, right bundle branch block), occasional chest x-ray findings, and definitive diagnosis on ventilation-perfusion scanning, spiral CT or pulmonary angiography.
Peripheral Resistance is caused by sepsis, anaphylaxis, or spinal shock
In sepsis, there are three fundamental physiologic upsets: increased synthesis of nitric oxide, activation of ATP-sensitive potassium channels in vascular smooth muscle, and deficiency of vasopressin. The plasma concentration of nitric oxide is markedly increased in septic shock. The production of this endogenous vasodilator appears to occur due to the expression of inducible nitric oxide synthetase by cytokines. This agent appears to be responsible for the end organ resistance to catecholamines and endothelin in sepsis.
If patient is awake, talking, and urinating, then hypotension is probably not shock
Look at lactate and base deficit
Shoot for MAP of 80 in normal folks, 90 in hypotensives
First look at the heart rate
Second look at volume status
Complete heart block, atrial fibrillation, tricuspid stenosis and regurgitation will lead to an inaccurate reading: although the diagnosis of these disorders can be made from the CVP waveform
The central venous pressure should be regarded as a trend. It is conventional to volume load an under-resuscitated patient to a target CVP: I use 8 10 mmHg if the non-ventilated patient, and 12 16 mmHg, if the patient is on positive pressure ventilation. If there is a question of cardiac disease, cardiac hypertrophy or dilatation or if the patient is middle aged or older, I aim higher 16 mmHg plus. In many young patients, it is often not possible to raise the CVP above 10 mmHg, such is the efficiency of the cardiovascular system.
Right atrial pressures are more representative of systemic vascular volume. Indeed with pulmonary hypertension, the use of left sided pressures may seriously overestimate the systemic blood volume. The purpose of PACs is to construct Starling (pressure-volume) curves of the left ventricle, to determine the end diastolic volume pressure relationship that optimizes stroke volume. The left ventricular end diastolic pressure is not measured directly, but through a surrogate the pulmonary capillary wedge pressure (PCWP).
Type
HR
SV
CVP
PCWP
CO/CI
PR
Hypovolemic
↑
↓
↓
↓
↓
↑
Distributive
Spinal Shock
↑
n
↓
↓
↑
↓
Anaphylaxis
↑
n
↓
↓
↑
↓
Sepsis
↑
↓
↓
↓
↑
↓
Cardiogenic
Heart Block
↓
↑
↑
↑
↓
Pump Failure
↑
↓
Relatively low
Relatively low
↓
↑
Vol Overload
↑
↓
↑
↑
↓
↑
Inflow obstruction
↑
↓
↑
↓
↑
Outflow obstruction
↑
↓
↑
↑
↓
↑
- There is no normal CVP or wedge pressures: you must follow a trend and look at responses to therapy, principally fluid boluses. Abnormal hearts those with acutely ischemic ventricles, fibrotic or contused myocardiums, are less compliant, and require higher filling pressures to project a normal stroke volume. They progressively dilate until there is sufficient muscle stretch to eject this volume. This is why the ejection fraction progressively falls. It is preferable to use stroke volume rather than cardiac output as the measured response to therapy, as the latter is influence by heart rate, which may be fast for a variety of reasons, and mask a poorly performing ventricle (in sepsis the cardiac output is characteristically high and the stroke volume low). A low mixed venous oxygen saturation (SvO2) is usually an indication of under-resuscitation, and volume loading is required. It is worthwhile to match this process with lactic acid concentration. An increased SvO2 is difficult to interpret: it may indicate inability to extract oxygen, it may indicate a hyperdynamic circulation. Normal hearts have huge physiologic reserve, and can cope very well with high filling volumes, consequently, you should never go wrong with aggressively volume loading shocked patients: you will adequately fill abnormal hearts without injuring normal hearts. Always aim your CVP and PCWP targets high! The first treatment for all kinds of shock, including cardiogenic, is volume, volume and more volume. A little extravascular lung water is probably less harmful than vasoactive drugs: do not skimp on the fluids for fear of flooding the lungs. There is no magic mathematical formula which allows you to figure out the influence of PEEP on PCWP and CVP: if transalveolar pressure is kept constant, the trend should be consistent. The pulmonary artery catheter is a monitor not a therapy: it is not a surrogate for resuscitative therapy or source control. During the resuscitative process, it should rapidly become apparent to you the CVP and PCWP that the patients heart likes (in terms of stroke volume and CVP). This is important with regard to fluid mobilization (deresuscitation) and redistribution. If the patients blood pressure or stroke volume begins to fall and the filling pressures are higher than those at which optimal stroke volume/cardiac output was obtained, then the patient has likely mobilized fluid, and is now overloaded: preload reducing agents such as nitrates and diuretics are required. An evolving increase in the stroke volume likewise represents hypervolemia: the patient will autodiurese if the kidneys are functioning normally. Be cautious with the use of derived variables from pulmonary artery catheters. I speak in particular of the systemic vascular resistance (SVR). There is, unquestionably, and abnormality in peripheral resistance (PR) in all forms of distributive shock, and appropriate vasoconstriction (and thus high PR) in hypovolemic and cardiogenic shock. Unfortunately, there is no easy method for measuring PR. The SVR is calculated from the equation for change in pressure/flow, which is MAP-CVP/CO. The problem with this equation is the pulsatility of cardiac output flow it is determined by the heart rate. A fast heart rate for the same stroke volume will lead to the measurement of a lower SVR than before. The PR may not have changed.
Determinants of cardiac output:The cardiac output is the product of the stroke volume and the pulse rate. It is calculated as:
CO = SV * PR.
The stroke volume of the left ventricle is ultimately determined by the interaction between its preload, the contractile state of the myocardium and the afterload that the ventricle faces. Unfortunately, there is no simple measure of the ‘contractile state’ and as a result, there is no single equation which describes the relationship between these three parameters.
Preload. That the ‘preload’ or stretch on myocardial fibres at the end of diastole had a significant effect on the subsequent force of contraction was recognised by the physiologist Otto Frank at the end of the nineteenth century 1 (Figure 1).
This fundamental relationship has since been analysed in great detail and the adjustment of preload by blood volume transfusion or depletion remains one of the most important therapeutic manoeuvres in acute cardiovascular medicine.
In practice, such volume adjustments can be made by various means:
1. Circulating blood volume can be increased by the administration of fluid, or reduced by the use of diuretics and / or fluid restriction.
2. Venous return can be varied by the adoption of a head-down or head-up posture.
3. Venous capacitance can be altered by the use of vasoconstrictor or vasodilator therapy.
Contractile State. In its strictest sense, the term ‘contractility’ refers to the inotropic state of the myocardium – that is, the force and velocity with which the myocardial fibres contract. This can be easily measured in an isolated muscle preparation under specified loading conditions, but is notoriously difficult to measure in the intact human.
In clinical practice, various contraction-phase indices such as velocity of fibre shortening, peak rate of ventricular pressure rise and end-systolic pressure:volume relationship (Figure 2) are used, but they are all affected by loading conditions to a greater or lesser degree.
The ‘chronotropic’ or ‘rate’ state of the intact heart should also be incorporated into any clinical definition of ‘contractility’ – because variations in the pulse rate can have obvious, important effects on the cardiac output and manipulation of pulse rate by the use of positive or negative chronotropes can be an important therapeutic manoeuvre in sick patients.
It is not possible to make any precise measurements of contractility with a PAC, although it is possible to make reasonable inferences about the contractile state by the use of ventricular function curves (Figure 1). This concept has been developed by Barash et al who have described the use of a ‘Hemodynamic Tracking System’ which defines the relationship between LVSWI and PAOP in patients with normal, slightly depressed and severely depressed ventricular function 2.
Adjustment of both the inotropic and chronotropic state of the heart by the use of inotropic drugs is commonly practised in cardiovascular medicine.
Afterload. In physiological terms, afterload can be defined as ‘The sum of all those forces which oppose ventricular muscle shortening during systole’ – although in a clinical sense it is probably more useful to consider systemic vascular resistance as the appropriate measure.
In isolated cardiac muscle, there is an inverse relationship between afterload and the initial velocity of shortening of the muscle (Figure 3). This would suggest a potential dependance of cardiac output on afterload. In fact, in the intact human, the output of the normal heart is relatively unaffected by changes in vascular resistance until afterload becomes quite extreme (Figure 4). This is probably because an increase in afterload leads to an almost immediate, secondary increase in preload by a ‘damming up’ of the blood within the left ventricle. This, in turn, increases end-diastolic volume which enhances contractility through the Frank-Starling mechanism. In contrast, if myocardial function is severely depressed, cardiac output may become crucially afterload-dependent as illustrated in Figure 4.
Thus, ‘sick’ hearts can be considered to be relatively preload independent but afterload dependent while the reverse is true for ‘healthy’ hearts. As a result, ‘afterload reduction’ (reduction of systemic vascular resistance by the use of appropriate vasoactive drugs) is of the greatest benefit in those in whom myocardial function is most depressed.
The role of blood viscosity and, indirectly, haemoglobin concentration in determining SVR is often overlooked. Although haemodilution is not commonly used as a therapeutic manoeuvre for afterload reduction, inadvertent haemodilution is often a concomitant of serious illness. Haematocrit and fibrinogen are the most important determinants of blood viscosity and, in turn, contribute significantly to vascular resistance. These relationships are illustrated graphically in Figure 5. Because blood is a non-Newtonian fluid, there is no simple expression to relate SVR to haematocrit and fibrinogen levels, however, it is easy to demonstrate the completely passive increase in venous return 3 and cardiac output which occur during haemodilution 4.
Eckmann et al have recently described the effect of variations in haematocrit and temperature on blood viscosity and have derived an equation which predicts blood viscosity as a function of temperature, shear rate, and haematocrit under a wide range of conditions 5.
Finally, it should not be forgotten that there is a degree of ventricular interdependence which can determine ventricular performance 6. – The position of the interventricular septum (IVS) can alter the compliance of each ventricle under altered loading conditions with secondary effects on contractility. This effect is not usually important, but can become so in conditions such as tension pneumothorax, tamponade, right ventricular infarction etc.
References:
1. Frank O: Zur Dynamik des herzmuskels. Ztschr fur Biol 32:370, 1895
2. Barash PG, Chen Y, Kitahata LM et al The Hemodynamic Tracking System. Anesth. Analg. 59:169 (1980)
3. Guyton AC, Richardson TQ Effect of hematocrit on venous return. Circ Res 9:157-163, 1961
4. LeVeen HH, Ip M, Ahmed N et al Lowering blood viscosity to overcome vascular resistance. Surg Gynecol Obstet 150:139-149, 1980
5. Eckmann DM, Bowers S, Stecker M, Cheung AT Hematocrit, volume expander, temperature, and shear rate effects on blood viscosity. Anesth Analg 2000 Sep;91(3):539-45
6. Taylor RR, Covell JW, Sonnenblick EH et al: Dependence of ventricular distensibility effect on filling of the opposite ventricle. Am J Physiol 218:711, 1967
Last edited on: 14/11/2000
Go to source: Determinants of cardiac output:
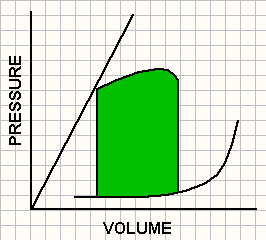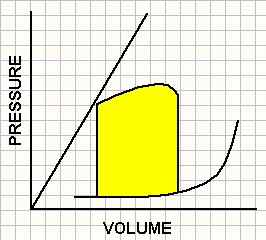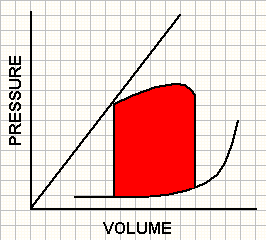
Pressure volume loops obtained in 3 contractile states.
A ventricle with increased contractility (green) operates at a lower end-diastolic volume and pressure and achieves end-systolic pressure at a lower end-systolic volume. In contrast, a ventricle with impaired contractility (red) operates at a high end-diastolic volume and pressure and achieves end-systolic pressure at a higher end-systolic volume.
The slope of a line drawn from the origin through the end-systolic pressure point is a measure of the contractile state.
Pressure:volume loops with end-systolic points on the same line are generated when loading conditions are changed, but contractility is unaltered.
This measure of contractility is thought to be relatively load-independent, but is very difficult to measure clinically.
Echocardiographic techniques for measuring similar indicators are emerging, but, as with many echocardiographic techniques, require considerable skill on the part of the observer.
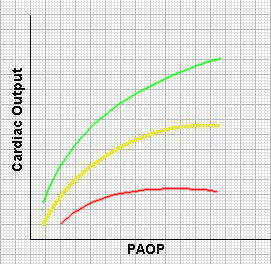
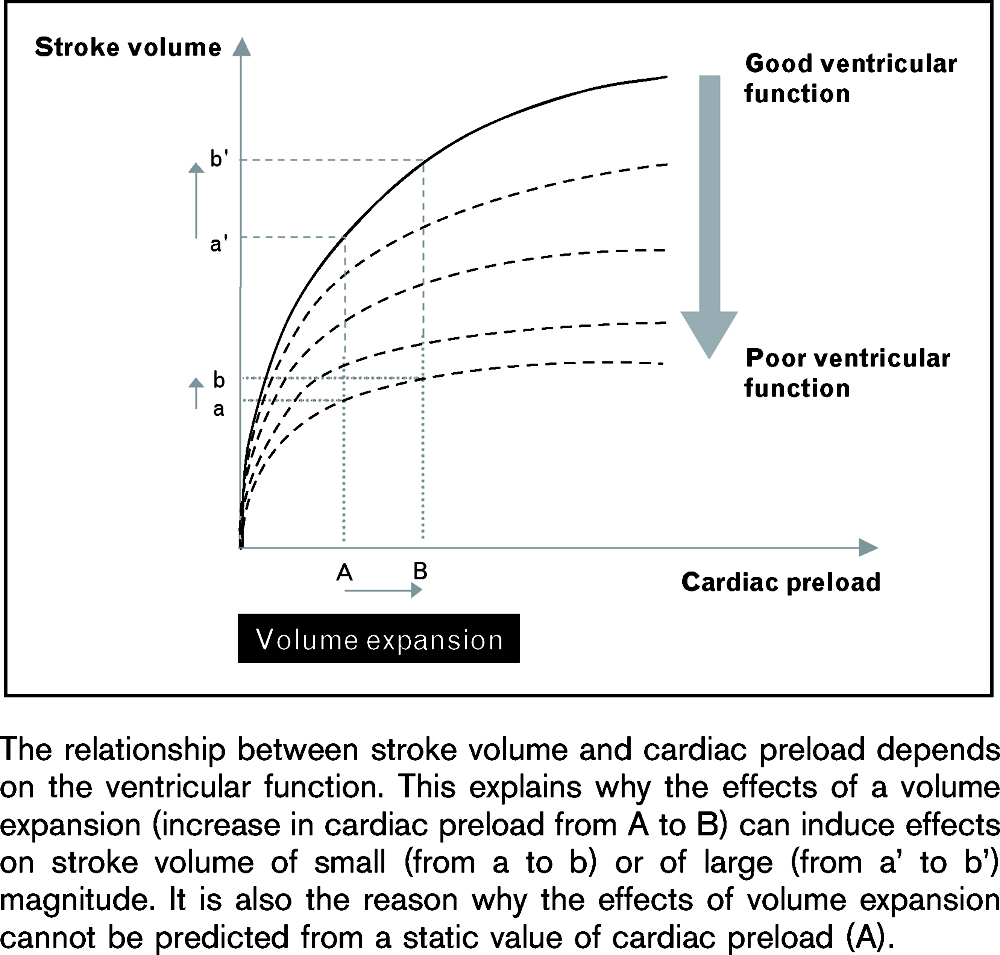
Ventricular function curves describe the fundamental Frank-Starling relationship. – As the amount of ‘stretch’ (preload) on the ventricular fibres is increased in diastole, so the resulting force of contraction of the next beat is increased. Note that in the failing heart (shown in red), the curve is relatively ‘flat’. Under these circumstances, increasing preload will not enhance ventricular performance. In fact, the reverse may occur because wall tension will increase with a concomitant increase in oxygen requirements of the heart.
The green curve represents a heart in which contractility is increased.
In this example, the cardiac output is used as an index of the force of contraction and the PAOP as a measure of sarcomere length.
Repeated measurements of PAOP and output can be made after therapeutic interventions such as volume loading or the use of inotropes. The results can be plotted onto a diagram of the form shown here and a notion of the ‘contractile state’ of the individual patient developed.
Endpoints of resuscitation
CVP=15
PAWP=20-22
CI>3
VO2>100cc/min/m2
Lactate<4
Base Deficit=-3 to 3
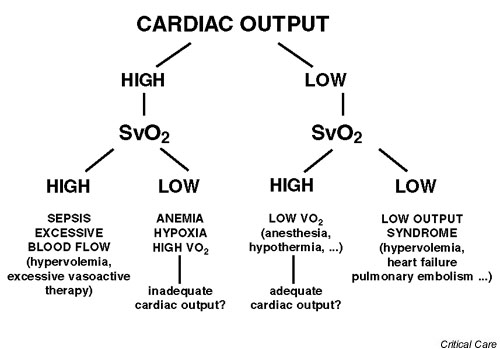
Pathophysiology
Insult causes decreased O2 delivery to cells (DO2). DO2 is the product of CO and PaO2. The body compensates by releasing epi and norepi which raise the CO. In addition, dopamine, cortisol, glucagon, growth hormone, and anti-diuretic hormone (ADH) are also released. Cells will increase the amount of Oxygen withdrawn from the blood causing decreases from the normal venous sat of 70-75%. When despite this increased extraction there is not enough oxygen, the cells begin anaerobic metabolism and release lactate. Eventually the acidosis surrounding the cells causes membrane disruption and cell death. Eventually, hypoxic cells of the vascular endothelium activate tissue macrophages and leukocytes, leading to the production of numerous harmful inflammatory mediators.7 Mediators that have been implicated in shock include tumor necrosis factor, interleukins 1 and 2, eicosanoids, interferon gamma, and platelet activating factor. With resuscitation, the hypoxic vascular beds are reperfused, resulting in the delivery of these mediators to the systemic circulation. It is this washout of inflammatory mediators that leads to the development of the systemic inflammatory response syndrome (SIRS). Due to compensatory mechanisms, the effects of age, or the use of certain medications, a large percentage of patients in shock will present with a normal blood pressure and heart rate. Thus, normal vital signs cannot be used to exclude the presence of shock. In fact, waiting for hypotension and tachycardia to develop to make the diagnosis invariably will increase patient mortality(EM Reports)
Base Deficit-amount of base that must be added to 1 liter of blood to normalize pH
MUST have peep on during all ventilations including BVM
In fact, strenuous accessory muscle use can increase consumption anywhere from 50% to 100%, leading to a decrease in cerebral blood flow by as much as 50%.3 Thus, the tachypneic patient in shock ultimately may require mechanical ventilation as respiratory muscles fatigue, hypercapnia increases, and acidosis worsens. Advantages to mechanical ventilation include improved oxygen delivery to the alveoli, correction of hypercapnia, and, most importantly, a decrease in oxygen consumption by the respiratory muscles.
Monitoring
CVP
Low: < 6 cmH2O;
Normal: 6-12 cmH2O;
High: > 12 cmH2O
JVD
In addition to noting simple engorgement, determine whether the veins remain distended or enlarge with inspiration. Normally, neck veins should collapse during inspiration, because of decreased intrathoracic pressure. In the presence of impaired venous return or elevated right heart pressures, neck veins will paradoxically swell (Kussmauls sign). This phenomenon occurs with tension pneumothorax, right ventricular infarction, pericardial tamponade, and massive pulmonary embolism.
Monitor by BP (Need mean of 60-65), Mental Status, Urine Output, and Base Deficit/Lactate
Dobutamine 5-20 ug/kg/min
Dopamine 5-20 ug/kg/min
Epinephrine 5-20 ug/min
Norepinephrine: 5-30 ug/min (0.2-1.3 mcg/kg/min)
Canadian Journal of Anesthesia 55:163-167 (2008)
Stability of norepinephrine infusions prepared in dextrose and normal saline solutions Conclusion: Norepinephrine solutions, in concentrations commonly used in the clinical setting, are chemically stable for seven days, at room temperature and under ambient light, when diluted either in D5W or NS.
Phenylephrine 2-200 ug/min
Auscultatory blood pressure measurement underestimates true arterial pressure in shock by an average of 15 mmHg, particularly when peripheral vascular resistance is high.21,22 However, Doppler measurements of blood pressure in hypotensive patients correlate well with direct arterial systolic blood pressure measurements
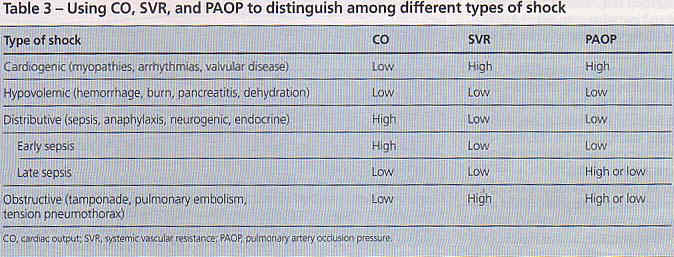
In non-crashing patients the MAP from arm cuff correlates with invasive monitoring; ankle and thigh is pretty good as well. Did not include circ collapse patients (Crit Care Med 2012;40(4):1207)
We are not good at detecting hypovolemia by physical exam. Postural tachycardia and dizziness are fairly good, rest are crap (Ann Emerg Med 2005;45(3):327)
Dobutamine
dobutamine is a racemic mixture. In a normal person with a low adrenergic state, the partial agonist and antagonist effects of the two enantiomers more or less balance out. In a high-adrenergic state, it is highly likely that *both* enantiomers will antagonise the effects of alpha agonists at the alpha receptor. My understanding is that the *affinity* of dobutamine for the alpha receptor is about 20 times that of noradrenaline (norepinephrine).
Norepinephrine
Canadian Journal of Anesthesia55:163-167 (2008)
Conclusion: Norepinephrine solutions, in concentrations commonly used in the clinical setting, are chemically stable for seven days, at room temperature and under ambient light, when diluted either in D5W or NS.
Multicenter RCT showed dopamine associated with greater adverse event rate than norepinephrine (NEJM 2010;362:779). Subgroup analysis showed dopamine increased the risk of death in cardiogenic shock patients. Most of the adverse events were dysrhythmias.
and Shock. 2009 Oct 21. [Epub ahead of print]
Efficacy and Safety of Dopamine versus Norepinephrine in the Management of Septic Shock.
Norepis decreases plasma volume by pushing fluid into interstium (as do prob all pressors) in septic shock (Acta Anaesthesiologica Scandinavica What is RSS?Volume 54, Issue 7, Pages 814-820)
Norepi actually increases preload and Cardiac Output (Critical Care 2010, 14:R142)
2nd study shows similar and qualified it with PLR (Crit Care Med 2011;39:689) Also supported by older studies (J Clin Invest 1957;36:1663; J Clin Invest 1959;38:1564; J Clin Invest 1965;44:1949; Anesth 2004;100:434)
Norepi caused improvement in NIRS measurement of microcirc (Int Care Med 2010;36:1882)
MA shows norepi is superior to dopamine for septic shock (Norepinephrine or dopamine for septic shock: systematic review of randomized clinical trials. J Intensive Care Med. 2012; 27(3):172-8 (ISSN: 1525-1489))
Epinephrine
Lancet. 2007 Aug 25;370(9588):676-84.Click here to read Links Norepinephrine plus dobutamine versus epinephrine alone for management of septic shock: a randomised trial. Annane D, Vignon P, Renault A, Bollaert PE, Charpentier C, Martin C, Troché G, Ricard JD, Nitenberg G, Papazian L, Azoulay E, Bellissant E; CATS Study Group. Raymond Poincaré Hospital (AP-HP), University of Versailles Saint Quentin, PRES UniverSud, Paris, France. djillali.annane@rpc.aphp.fr BACKGROUND: International guidelines for management of septic shock recommend that dopamine or norepinephrine are preferable to epinephrine. However, no large comparative trial has yet been done. We aimed to compare the efficacy and safety of norepinephrine plus dobutamine (whenever needed) with those of epinephrine alone in septic shock. METHODS: This prospective, multicentre, randomised, double-blind study was done in 330 patients with septic shock admitted to one of 19 participating intensive care units in France. Participants were assigned to receive epinephrine (n=161) or norepinephrine plus dobutamine (n=169), which were titrated to maintain mean blood pressure at 70 mm Hg or more. The primary outcome was 28-day all-cause mortality. Analyses were by intention to treat. This trial is registered with ClinicalTrials.gov, number NCT00148278. FINDINGS: There were no patients lost to follow-up; one patient withdrew consent after 3 days. At day 28, there were 64 (40%) deaths in the epinephrine group and 58 (34%) deaths in the norepinephrine plus dobutamine group (p=0.31; relative risk 0.86, 95% CI 0.65-1.14). There was no significant difference between the two groups in mortality rates at discharge from intensive care (75 [47%] deaths vs 75 [44%] deaths, p=0.69), at hospital discharge (84 [52%] vs 82 [49%], p=0.51), and by day 90 (84 [52%] vs 85 [50%], p=0.73), time to haemodynamic success (log-rank p=0.67), time to vasopressor withdrawal (log-rank p=0.09), and time course of SOFA score. Rates of serious adverse events were also similar. INTERPRETATION: There is no evidence for a difference in efficacy and safety between epinephrine alone and norepinephrine plus dobutamine for the management of septic shock.
Epi as an Inotrope
This how it is used in CTICUs
0.01-0.07 mcg/kg/min for CI at leas 2.2
Dopamine
Greet van den Berghe’s work shows neuroendocrine dysfunction as well as immunological modulation secondary to prolactin
(Anesth & Analg, 2004, 98,461-468)
Reasons Dopamine is Bad
Does not benefit the renal system
Induces Natriuresis and Diuresis
Shunts blood away from outer medulla, which is the region most prone to ischemic damage
Possible induction of decreased splanchnic perfusion
Decreases GI Motility
Impairs ventilatory response to hypoxemia and hypercapnia
Effects on anterior pituitary–decreases prolactin secretion
and (Holmes CL and Walley KR, Bad medicine: low-dose dopamine in the ICU, Chest, 2003, 123: 1266-1275)
Meta-Analysis: Low-Dose Dopamine Increases Urine Output but Does Not Prevent Renal Dysfunction or Death Jan O. Friedrich, MD, DPhil; Neill Adhikari, MD, CM; Margaret S. Herridge, MD, MPH; and Joseph Beyene, PhD Ann Intern Med 5 April 2005 | Volume 142 Issue 7 | Pages 510-524 Renal dose also causes a. fib in post op pts (Crit Care Med 2005;33:1327)
Assoc c higher mortaility in Sepsis patients in the SOAP study:
Conclusions: This observational study suggests that dopamine administration may be associated with increased mortality rates in shock. There is a need for a prospective study comparing dopamine with other catecholamines in the management of circulatory shock. (Crit Care Med 2006;34(3):589)
Gastric tonometry
measures pH of cells lining gut; based on hypothesis that CO2 in lumen of stomach or intestine in rapid equilibrium with CO2 in cells lining them; during ischemia, cells switch to anaerobic metabolism, and PCO 2 increases; splanchnic circulation believed to be first to develop ischemia; basing management on restoring gastric intramucosal pH more effective in lowering mortality than management based on hemodynamic parameters; difficult to perform; more practical new development may be noninvasive sublingual tonometry
Trendelenberg
No evidence that it works, may make things worse (Canadian JEM Vol. 6, No. 1, January 2004)
trendelenberg does not work as shock maneuver 24. Sing R, O’Hara D, Sawyer MJ, et al. Trendelenburg position and oxygen transport in hypovolemic adults. Ann Emerg Med 1994;23:564568.Bibliographic LinksMount Sinai Serials 25. Taylor J, Weil MH. Failure of Trendelenburg position to improve circulation during clinical shock. Surg Gynecol Obstet 1967;122:10051010.Mount Sinai Serials 26. Bivins HG, Knopp R, dos Santos PAL. Blood volume distribution in the Trendelenburg position. Ann Emerg Med 1985;14:641643.Bibliographic LinksMount Sinai Serials 27. Gaffney FA, Bastian BC, Thal ER, et al. Passive leg raising does not produce a significant auto transfusion effect. J Trauma 1982;22:190193.Ovid Full TextBibliographic LinksMount Sinai Serials
EMJ BETS (EMJ 2010;27:877) It doesn’t do squat.
Three systems respond to hypotension:
sympathetic
vasopressin
renin/angiotensin
If two are blocked then difficult to control hypotension
may need Vasopressin 3-5 unit IV bolus if other two systems are blocked
Push Dose Pressors
Phenylephrine
Comes in 1% 1cc vials this means 10 mg/cc
Dilute in 10 cc syringe and you get 1 mg/cc
Dilute again 1 cc in 10 cc and you get 100 mcg/cc
give 50-100 mcg (.5-1 cc at a time) at a time
max 1 mg/dose)
then 40-60 mcg/min
potent, rapid onset, short duration
Ephedrine
Comes in 50 mg 1 cc vial; dilute into 10 cc of NS to get 5 mg/cc
5-25 mg IV q 5-10 min
primarily direct beta, with some indirect alpha
large doses required
slow onset, long duration
Peak pressor effect of phenyl 61.8 sec, peak decreased CO 30 sec
Ephed Peak pressor 90, peak CO 58.8
equivalence for map ~125:1
(Anesth 2009;111:753)
Calcium
100 mg then 1-5 mg/hr
synergistic with vasopressors, increased MAP, increased SVR, increased inotropy
Hemodynamic effects of ca as bolus and drip (Annals of Thoracic SurgeryVolume 37, Issue 2, February 1984, Pages 133-140)
iCal directly correlates with arterial pressure (Crit Care Med 1988;16(6):578)
A randomized, blinded, placebo-controlled evaluation of calcium chloride and epinephrine for inotropic support after emergence from cardiopulmonary bypass. (Anesth Analg. 1992 Jan;74(1):3-13)
Obstructive Shock
Decreased Diastolic Filling
Tamponade
Pneumothorax
Increased Ventricular Afterload
Pulmonary embolism
use norepi (amrinone promising, but not enough studies (Emedhome.com article.)
Cardiogenic
Cardiogenic Shock
For SBP > 80 mmHg, dobutamine is recommended as the initial agent of choice. It has been shown to cause less tachycardia, vasoconstriction, and arrhythmia than other agents (38,39,40). Additionally, dobutamine increases coronary blood flow and collateral blood flow to ischemic areas while raising myocardial contractility and cardiac output, but lowering left ventricular filling pressures (ref 39,41).
For Moderate Hypotension (e.g. SBP < 80 mmHg), dopamine is recommended as the agent of choice since vasoconstriction of peripheral vessels is needed to maintain vital organ perfusion.
For Profound Hypotension (e.g. SBP < 70 mmHg) or refractory hypotension, norepinephrine is recommended. Hemodynamic studies of acute myocardial infarction in cardiogenic shock treated with norepinephrine have shown a rise in mean arterial pressure and systemic vascular resistance. These studies also revealed improved myocardial perfusion and oxygenation, however there was no change in cardiac output. This failure to augment cardiac output is thought to represent the magnitude of the ischemic zone and lack of inotropic reserve (ref 42). The phosphodiesterase inhibitors, amrinone and milrinone, are known to increase contractility. They do not stimulate adrenergic receptors and play a reserve role when other catecholamines are ineffective, or when beta receptors are blocked.
Thrombolytics, due to poor reperfusion rates, have shown no mortality benefit in cardiogenic shock in large, randomized studies (ref 43,44). Intra-aortic balloon pumps decrease systemic afterload and increase diastolic perfusion pressure without increasing oxygen demands. While balloon pump use is considered standard of care, no improvement in outcome has been associated solely with its use. However, IABP does seem to function as a bridging device to revascularization. The ongoing TACTICS (thrombolysis and counterpulsation to improve cardiogenic shock survival) will address the role of IABP as an adjunct to thrombolysis (ref 45).
Vasodilating agents will increase CO, decrease PCWP; with little change in art pressure
at moderate and high doses there will be a decreased ABP
The increase in CO is seen in patients with high LV filling pressures, in normal patients the loss of filling will decrease CO b/c of decreased starling. (Circulation 1973;48:1183)
Cardiogenic Shock needs early revascularization (JAMA 2006;295(21):2511)
Review Article (Curr Opin Crit Care 2006;12:431)
SHOCK trial (NEJM 1999;341:625)
cardiac power important prognostically: product of CO and MAP
Effect of Vasopressin in card shock from MI (Am J Cardiol 2005;96:1617)
vasopressin, in studies of septic shock, has been shown to increase MAP without affecting PCWP or CO
norepi increased cardiac power; vasopressin did not
retrospective and most patients on multiple pressors
Review of Inodilators (Am J Cardiol 2005;96(supp):47G)
Milrinone and dobutamine seem to have similar outcomes and side effects
In advanced vasodilatory shock, disease oriented outcomes may be better with norepi+vaso when compared to norepi alone (Circ 2003;107:2313)
horrible case series of inadequate pressor doses, then pts had vasopressin added. In 2 of the 3 cases CI then decreased. (Crit Care Med 2000;28(1):249)
review article (Med Clin N Am 2007;91:713)
Study of vaso vs. norepi for hyptension after milrinone showed vaso had less effects on PVR, but the MAPs were driven higher with norepi confounding the study (Eur J Cardio-Thor Surg 2006;29:952)
Editorial Letter pointing out problems with this study (Eur J CT surg 2006;30:686)
New study may indicate benefit from pushing MAPS higher (Shock. 2014 Apr;41(4):269-74. doi: 10.1097/SHK.0000000000000099. Increasing mean arterial pressure in cardiogenic shock secondary to myocardial infarction: effects on hemodynamics and tissue oxygenation.)
Right Heart Failure

norepi/dob vs. epi; epi has more lactic acidosis, and dysrhythmia so they recommend the former (Crit Care Med 2011;39:450)
Hypothermia
Mild ther. hypothermia increased SVR, improved hemodynamics, and reduced oxygen consumption (Crit Care Med 2012; 40(6):1715)
Milrinone
Am Heart J. 2001 Feb;141(2):266-73.Pharmacodynamic effects of milrinone with and without a bolus loading infusion.Baruch L, Patacsil P, Hameed A, Pina I, Loh E.Bronx Veterans Affairs Medical Center, Bronx, NY, USA. BACKGROUND: Milrinone is a positive inotropic agent with vasodilatory and lusitropic activity. Milrinone dosed as a 50 microg/kg bolus followed by a continuous infusion provides an immediate and sustained hemodynamic response. The comparative pharmacodynamics of a placebo bolus and a milrinone bolus followed by a continuous milrinone infusion in patients with decompensated heart failure are unknown. METHODS: Nineteen patients with decompensated heart failure underwent right heart catheterization and were randomized to receive an intravenous infusion of milrinone at a rate of 0.50 microg/kg/min with (n = 9) or without (n = 10) a preceding 50 microg/kg bolus. Pulmonary capillary wedge pressure, cardiac index, and plasma milrinone levels were measured serially over 24 hours. RESULTS: In the milrinone bolus group, maximal effects on plasma concentration (352.3 ng/mL), cardiac index (+0.97 L/min/m(2), P =.02), and pulmonary capillary wedge pressure (-11.25 mm Hg, P <.001) were seen after the loading dose. In the placebo loading dose group, significant hemodynamic effects were observed starting at 30 minutes after the start of the continuous infusion. Changes in pulmonary capillary wedge pressure (placebo -8.6 vs milrinone -8.78 mm Hg, P not significant [NS]) were similar in both groups at 2 hours, whereas changes in cardiac index (placebo loading +0.81 vs milrinone loading +0.78 L/min/m(2), P NS) and milrinone levels (placebo loading 168.0 vs milrinone loading 165.6 ng/mL, P NS) were similar at 3 hours. One patient randomized to a milrinone bolus demonstrated a marked decrease in blood pressure and was discontinued from therapy. CONCLUSIONS: A milrinone infusion without a bolus appears to be a rapidly effective inotropic strategy that may have an improved safety profile during the initiation of therapy compared with a continuous infusion strategy initiated with a bolus.
Tachycardia-related Hypotension
TCA overdose
Norepinephrine
new class includes calcium sensitizer levosimendan (Simdax), opens potassium channels in vascular smooth muscle for vasodilator effect, long-term use not associated with increased mortality, in contrast to other inotropic agents; significantly improves cardiac function, ejection fraction, and functional status
Hypovolemic
Sources Of Occult Hemorrhage.
Trauma
- Solid organ injury
- Pulmonary parenchymal injury
- Myocardial laceration/rupture
- Vascular injury
- Retroperitoneal hemorrhage
- Pelvic fracture
- Ruptured duodenum
- Ruptured kidney
- Fractures, especially bones and pelvis
- Lacerations, especially scalp
- Epistaxis
Gastrointestinal tract
- Mallory-Weiss tear
- Esophageal varices
- Peptic ulceration
- Gastritis/esophagitis
- Meckels diverticulum
- Malignancies
- Vascular lesions (arteriovenous malformations, rare congenital anomalies)
- Inflammatory bowel disease
- Ischemic bowel disease
- Diverticular disease
Reproductive tract
- Vaginal bleeding
- Malignancies
- Miscarriage
- Metrorrhagia
- Retained products of conception
- Placenta previa
- Vaginal or uterine laceration
- Ectopic pregnancy
- Ruptured ovarian cyst
Vascular
- Aneurysms
- Dissections
- Arteriovenous malformations
- Vasculitis (e.g., Henoch-Schönlein purpura)
Others
- Tumor
- Infection (e.g., Mycobacterium tuberculosis [Rasmussens aneurysm])
- Spontaneous splenic rupture
- Bleeding dyscrasias
One final controversy in the treatment of patients in hemorrhagic shock is the addition of antioxidants and/or free radical scavengers to resuscitative fluids. Hemorrhagic shock impairs antioxidant defense mechanisms and increases free radical production.22 As discussed in detail above, free radical formation plays a key role in the pathogenesis of shock and subsequent MODS. To date, the results of studies involving superoxide dismutase, N-acetylcysteine, ascorbic acid, vitamin E, and even deferoxamine have been published.23 Preliminary data imply that the inflammatory response to shock is somewhat mitigated by the addition of an antioxidant to the resuscitation fluid
hydrocortisone increased sensitivity to catecholamines after resus from hemorrhagic shock (crit care med 2005;33(12):2737)
Distributive
Neurogenic Shock
Dopamine or (ephedrine and atropine)
Anaphylaxis
EPI, final dilution of 1:100,000 infused over 5 to 10 minutes by mixing 0.1 mg (0.1 ml) of 1:1000 with 10 ml of normal saline. This is equivalent to a 100 mcg bolus given at 10 mcg/min. Once therapy has begun, a continuous infusion could be delivered with 0.5 to 5 mcg/min titrated to clinical response
Septic Shock
Start c Norepi until MAP>65 then add dobutamine. (EMEDHOME.com Article JB18)
Septic Shock Systemic inflammatory response syndrome (SIRS): by original definition, almost everyone in ICU has SIRS; new definitions may stage SIRS by genetic abnormalities predisposing patient to sepsis, degree of infection, response to infection based on mediators and markers, and presence of organ dysfunction Antibiotic therapy: choice depends onorganism and site of infection; community-acquired vs hospital-acquired infection; host factors, eg, immunosuppression; severity of infection; local patterns of antibiotic susceptibility and resistance; begin with broad-spectrum antibiotic, then narrow choices based on culture and sensitivity; follow published guidelines for treatment of community-acquired and ventilator-associated pneumonia Hemodynamic therapy: preload, afterload, and contractility different in sepsis; pressure (eg, wedge pressure) and volume (left ventricular end diastolic volume) related by ventricular compliance; in sepsis ventricular compliance increased, ventricle more relaxed and has higher filling volume than expected at given wedge pressure or central venous pressure; goal is optimized ventricular filling; whereas in other conditions, optimal cardiac output occurs at wedge pressures of 15 to 18 mm Hg, in sepsis, it occurs at lower pressures, eg, 5 to 15 mm Hg, that cannot be predicted; give fluid boluses until hemodynamic improvement ceases, regardless of wedge pressure, which may be only 5 mm Hg; contractilitymay be markedly decreased in spite of high cardiac output caused by tachycardia; due to optimized preload, end diastolic volume increased; stroke volume normal, so ejection fraction decreased; afterload also decreased, indicating marked decrease in contractility; not due to development of myocardial ischemia; down-regulation of beta- adrenergic receptors may play role but, main mechanism thought to be production of myocardial depressant factors, eg, tumor necrosis factor and interleukin-1 Inotropic therapy: dobutamine5 to 20 µg/kg per min; has beta-adrenergic effect, ie, increases cardiac output, heart rate, and stroke volume; decreases SVR from vasodilation, has variable effect on BP; if BP declines, give fluid challenges; dopamine2 to 20 µg/kg per min; increases cardiac output by increasing stroke volume rather than heart rate; does not appear to increase SVR; has variable effect on splanchnic circulation; no evidence for renal dose dopamine that would improve renal blood flow; epinephrine20 to 300 ng/kg per min; has beta-adrenergic effects at lower, doses with resulting increase in BP due to increased cardiac output; alpha-adrenergic effects at higher doses cause BP increase due to increased SVR; worsens lactic acidosis and decreases splanchnic blood flow, particularly in combination with norepinephrine; norepinephrineused as afterload agent; raises BP and maintains tissue perfusion in septic shock with optimized hemodynamic circulation and elevated cardiac output, as opposed to effects in hypo-volemic and cardiogenic shock (decreased cardiac output, renal failure, bowel ischemia); increase in BP in septic shock due mainly to SVR (heart rate and cardiac output do not change); glomerular filtration rate and urine output improve when vasoconstrictors used in high-output septic shock, renal blood flow maintained; norepinephrine used alone tends to improve splanchnic blood flow, but response variable; definitely improves splanchnic blood flow in combination with dopamine or dobutamine; phenylephrinedata suggest benefits similar to norepinephrine in septic shock; vasopressindoes not constrict renal vasculature; dilates cerebral, coronary, and pulmonary vasculature; small studies suggest vasopressin stabilizes or improves BP, improves urine output and creatinine clearance; used as second-line therapy in septic shock when standard drugs cannot maintain BP without risking adverse renal effects Steroid therapy: low-dose steroids postulated to address adrenal insufficiency in septic shock; study datarandomized patients to receive placebo or 100 mg hydrocortisone q8h for at least 5 days; greater reversal of shock at 7 days and trend toward reduced mortality among steroid-treated patients; study of similar regimen with dosages individualized by weight showed earlier discontinuation of vasopressor therapy with hydrocortisone (2 days vs 7 days); third study enrolled 310 patients on mechanical ventilation; all received corticotropin-stimulation test, then were randomized to receive 50 mg hydrocortisone q6h or placebo; response to corticotropin-stimulation test defined as increase >9 µg/dL; steroid-treated patients had increased reversal of shock, trend toward improved 28-day survival; benefit limited to patients unresponsive to corticotropin-stimulation test (responders do not have adrenal insufficiency) Activated protein C: complex anticoagulant, anti-inflammatory compound; levels decreased in sepsis and decrease correlates with outcome; Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trialrandomized 840 patients to receive placebo or 96-hr infusion of 24 µg/kg per hr recombinant human activated protein C (drotrecogin alfa, Xigris); mortality reduced to 25% with Xigris compared to approximately 31% with placebo; effect seen primarily in patients with APACHE II scores <25; significant improvement in mortality among sickest 50% of patients; bleeding complications increased from 2% in placebo group to 3.5% in Xigris-treated patients (primarily patients who developed thrombocytopenia or those undergoing invasive procedures without discontinuation of Xigris) Trends in mortality from septic shock: mortality approximately 65% prior to modern critical care; now approximately 40%, with many studies showing mortality in 30% range; further decline expected over next 5 yr (Audiodigest Anesthesiology)
Vasopressin
indicated for catecholamine resistant septic shock. Dose should be no greater than 0.04 U/min to avoid side effects on heart and splanchnic vasculature.
(0.010.04 units/min)
Best review article (Volume 30 Number 7 of Intensive Care Medicine)
Review (Inten Care Med 2004;30:1276)
Vasopressin levels are high in early shock and then at 24 hrs begin to become deficient (Crit Care Med 2003;31(6):1752)
Decreased gut perfusion c vaso (Systemic Effects of Vasopressin Administration Oral Presentation 14th Annual Congress S138)
CI impaired with terlipressin compared to norepi (Crit Care Med 2005;33(9):1897)
Review (Chest 2001;120:989)
Review of vaso and terli (Crit Care 2005;9(2):212)
Vaso and Norepi vs. Norepi in under-resused pts showed slightly better creat clearance in vaso group (Anesthesiology 2002;96:576)
another review that states vasopressin does not vasoconstrict the pulmonary vasculature, while norepi may. Also potentially better effects on the kidney. (Can J Anesth 2006;53(9):934)
RCT of norepi vs. vasopressin, aso was ineffective in patients eve at doses beyond the safe limit (Inten Care Med 2006;32:1782)
Vasopressin does not cause as much pulmonary vasoconstriction (Br J Anaesth 2007;99(4):552)
Vasst (NEJM Volume 358:877-887 February 28, 2008 Number 9 Next Vasopressin versus Norepinephrine Infusion in Patients with Septic Shock)
Addisonian Shock
Steroid Insufficiency

Shock in Trauma
Hemorrhagic
External Chest Abdomen Pelvis Long bone
Blood volume = 7% of weight in kg (70 cc/kg)
Non-Hemorrhagic
Tension Pneumothorax Pericardial Tamponade Myocardial Contusion Spinal Shock
In head injury patients, norepinephrine is probably better than dopamine (CCM 2004 32:4)
Effects of Acidosis
Title Effects of acidosis and hypoxia on the response of isolated ferret cardiac muscle to inotropic agents. Source Cardiovascular Research. 28(8):1209-17, 1994 Aug. Local Messages 1990-1996 Abstract OBJECTIVE: The aim was to study the effects of acidosis and hypoxia on the response of cardiac muscle to inotropic agents which (a) act predominantly by increasing intracellular [Ca2+] (raising extracellular [Ca2+], noradrenaline, isoprenaline) and (b) act partly (phenylephrine) or predominantly (EMD 57033) by increasing myofilament calcium sensitivity. METHODS: The experiments were performed on isometrically contracting, isolated ferret papillary muscles (n = 45). For each intervention dose-response curves were performed in control solution (pH 7.35), in hypercapnic acidosis (pH 6.85), and in hypoxia (produced by replacing O2 with N2 in the superfusing solution). In some experiments, the photoprotein aequorin was microinjected into superficial cells of the preparation in order to measure intracellular [Ca2+] as well as force. RESULTS: The results were broadly similar for both classes of inotropic agent. Acidosis caused a shift of the pCa-tension curve to the right (desensitisation of the myofilaments to calcium), but had no significant effect on maximum force. A sufficient inotropic stimulus supplied by either class of inotropic agent could completely reverse the negative inotropic effects of acidosis. The main difference between the two inotropic mechanisms was that the enhanced force produced by calcium sensitisers was associated with a reduction in calcium transient amplitude, while the other inotropes increased the amplitude. The main effect of hypoxia was to decrease maximum force. All the inotropes tested were relatively ineffective in reversing the force depression due to hypoxia. CONCLUSIONS: The negative inotropic effects of acidosis can be reversed by a sufficiently large inotropic stimulus. Since calcium transient amplitude is already increased in acidosis, the results suggest that calcium sensitisers are likely to be less arrhythmogenic in this situation. The relative ineffectiveness of the inotropes in hypoxia indicates that the main mechanisms causing reduced force in this situation lie downstream of the mechanisms of action of the inotropic agents tested.
Mild acidosis (7.2) impairs the hearts response to beta-adrenegic stimulation (Crit Care 2012;16:R153)
MA of hemodynamic optimization optimization of tissue perfusion showed decreased mortality (Crit Care Med 2002;30(8):1688)
Persistent microcirc abnormailities assoc c death nd organ faiure. used subingual co2 (crit care med 2004;32(9):1825)
Review of targeted resus after trauma (Curr Opin Crit Care 2004;10:529) critical o2 delivery is the key steal figure talks about rvedv swan, pcco, esoph doppler 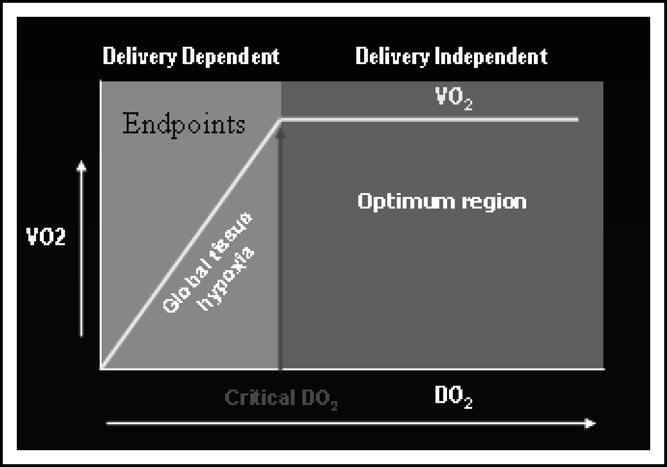
Low-Tech Management of Shock
Unified theory of shock
It all comes down to end-organ perfusion
Is intravascular volume (preload) adequate?
Is blood flow adequate?
Is vascular resistance appropriate?
Is oxygen transport balance adequate?
Using PAOP assumes that ventricular compliance is unchanging and pressure reflects end diastolic volume
Preload~LVEDV (unless ventricular geometry is changed)
LVEDV~LVEDP (unless ventricular compliance is changed)
LVEDP~LAP (unless there is mitral disease, or intrathoracic or intrabdominal pressure changes)
LAP~PAOP (unless catheter is malpositioned or intrabdominal/intrathoracic pressures are elevated
Coronary Perfusion Pressure=DBP-PAOP
Must maintain over 50 mmHg
Abdominal Perfusion Pressure=MAP-IAP
Should maintain over 50
CO=SVI x HR
Vascular resistance
SVRI (dynes*sec*cm -5)=(MAP-CVP)80 / CI
PVRI=MPAP PAOP 80/CI
Causes of increased pvri
Pulmonary htn, ards, intra-abd htn, mitral stenosis, aortic stenosis, left heart failure
Ventricular stroke work indices
Work=force x distance or change in pressure x change in volume
LVSWI=(MAP-PAOP) (SVI) (0.0136) (g m/m2)
RVSWI=(MPAP-CVP) (SVI) (0.0136) (g m/m2)
Decreased by inadequate volume, increased resistance, or decreased contractility
Increased by ventricular hypertrophy or physiologic conditioning
CCO
Need volumetric data
RVEF=right ventricular ejection fraction
RVEDI=right ventricular end-diastolic volume index
Independent of zero-pressure references and changing compliance
Proof: Cheathem et al. 2000
RVEDI=CI / (HR RVEF) or = SVI/RVEF
Incorrect placement, mitral valve disease, or irregular heart rate can still screw up the measurements
RVEDI reflects preload status
RVEF reflects contractility and afterload
RVEF
Normal Pt
Crit Ill Pt
.2
200
240
.3
150
180
.35
125
150
.4
100
120
.5
50
60
Oxygen Transport
Is tissue oxygen delivery sufficient to meet cellular oxygen demand?
DO2=Delivery-oxygen pumped to the tissues by the heart
VO2=Consumption-amount of oxygen consumed by the tissues
Oxygen Demand=the amount of oxygen required by the tissues to function aerobically. May exceed consumption and delivery in critical illness
Four new Questions:
Does oxygen delivery meet the patients needs?
Is cardiac output adequate for consumption?
Is oxygen consumption adequate for demand?
Is patients hypoxemia due to a pulmonary problem or to a low flow state?
Supply and extraction must meet demand
If demand exceeds supply then shock is present
When consumption just meets demand patient is on the brink of decompensation
You need physiologic oxygen reserve
Oxygen content=oxygen bound + oxygen dissolved
C*O2=(1.34x Hb x S*O2)+(P*O2x0.0031)
Where * signifies the location
Each g of hemoglobin can carry 1.34 ml of O2
The solubility of oxygen is 0.0031 cc/dL
The saturation of Hb varies depending on the FiO2 and the presence of mixed, unoxygenated blood
The oxygen content of the blood as it leaves the alveolus is:
(assuming saturation of 1.0 as long as FiO2>0.21)
Pb=PAO2 +PACO2 + PAN2 + PH2O
PAO2=FiO2 x ((PB-PH20)-(PaCO2/RQ))
Where PB=barometric pressure
PAO2=FiO2 x ((760 torr- 47 torr)-(PaCO2/0.8)
Can be approximated by FiO2 X (650)
Pulmonary End-Capillary Oxygen Content (CcO2)
CcO2=(1.34xHbx1) + (PAO2 x 0.0031)
Normally about 20.7
PAO2 normally = 200 at 30%
CaO2=(1.34 x Hb x SaO2) + (PaO2 x 0.0031)
=20.1 cc/dL + 0.31 cc/dL
=20.4
CvO2=1.34 x Hb x SvO2 + PvO2 x 0.0031
Can estimate PvO2 at 35 with fairly high accuracy
(1.34 x 15 x 0.75) + (35 x 0.0031)
=15.1 + 0.11
=15.2 cc / dL
Ca-vO2=CaO2 CvO2
=20.4-15.2
=5.2 cc/dL
Oxygen Delivery Index
DO2I=CI x CaO2 x 10 dL/L
=4 x 20.4 x 10
=800 cc O2/min-m2
Oxygen Consumption Index
Volume of O2 returned to the right atrium
VO2I=CI x Ca-vO2 x 10
=4 x 5.2 x 10
=~200 cc O2/min-m2
Oxygen extraction ratio or utilization coefficient
Percentage of delivered oxygen which is consumed by the body
OUC=VO2I/DO2I
=~0.25
If SaO2>0.92 then OUC can be estimated as 1-SvO2
Mixed venous is the flow-weighted average of the venous saturations of the all of te venous beds
If SvO2 decreases then consumption is up or delivery is down
The four determinants are Hb, CI, SaO2, oxygen consumption index (VO2I)
Is DO2I adequate for the patients needs?
Should be at least 10 cc/kg/min
If it is less, check SvO2 or OUC
>0.35 suggests that needs are barely being met
Ensure that Hb is appropriate
When delivery exceeds demand then VO2I wil plateu and no longer rise in response to DO2I
When it is insufficient, there is supply dependency, this is the critical DO2I
Is CI adequate for VO2I
Check the Ca-vO2
Check the heart rate and stroke volume
If it is less than 5, then CI is sufficient to meet the bodys demands
If it is >5, then an abnormally high percentage of oxygen is being extracted
Attempt to increase DO2I and CI
Is consumption adequate for demand
Check lactate level, If>2.0,
Is hypoxemia due to pulmonary problem or to a low flow state
Calculate the shunt fraction aka the venous admixture
Normal shunt is 2-5%
May exceed 50% in patients with ARDS
Commonly estimated by Aa gradient
Sources of normal shunt
Bronchial artery which enters pulmonary veins
Desaturated blood from Thebesian veins after perfusing myocardium
Normal alveolar collapse in Wests Zone I at the Apices
Abnormal sources of pulmonary shunt
Atelectasis
Lobar pneumonia
Inhalation
Drowning
ARDS
Abdominal Compartment Syndrome
Qt=total cardiac output
Qs=shunted portion of the cardiac output
Qns=normal pulmonary end-capillary blood flow that is not shunted past abnormal alveoli
Qt=Qs+Qns
Qt=CaO2=total oxygen delivered to the body
Qs=CvO2=total oxygen within the shunted blood
Qns=CcO2=total oxygen within end capillary blood
Qt(CaO2)=Qs(CvO2)+Qns(CcO2)
Qs/Qt=(CcO2-CaO2)/(CcO2-CvO2)
Low Tech Management of Shock
Endpoints
No advantage to MAP of 85 over 65 (Crit Care Med 2005;33(4):780)
Opening the microcirculation
(Intens Care Med 2002;28:1208)
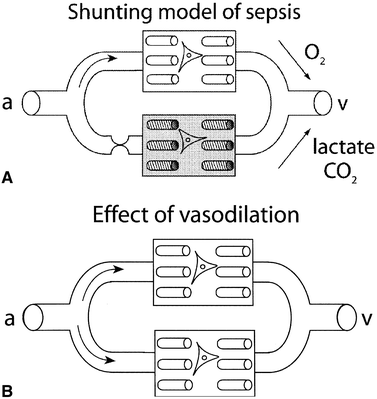
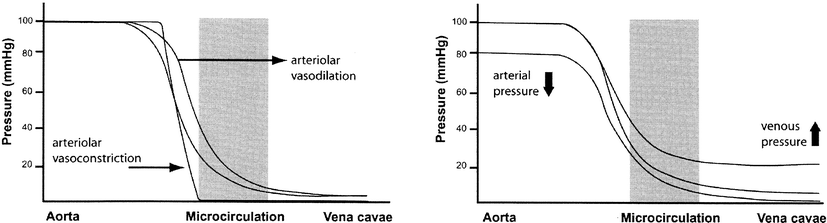
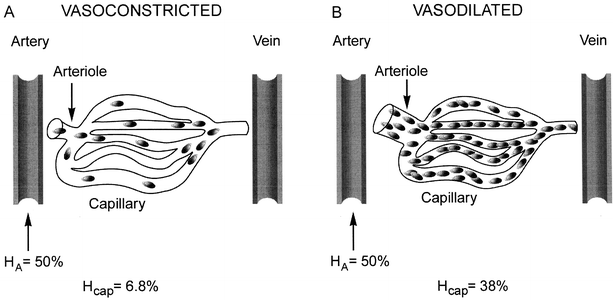
continued elevation of indicators of tissue dysoxia may actually represent cytopathic hypoxia, a problem of oxygen utilization by the mitochondria
but can also be due to microcirculatory shunting
PGI2 can cause dramatic increases in oxygen consumption demonstrating recruitment of microcirculation
Bench to Bedside Review (Crit Care 2006;10:221)
In sepsis, all the components of the microcirculation listed above are affected, causing a severe dysfunction in its regulatory function and resulting in a regional mismatch of oxygen supply and demand [4]. In summary, endothelial cells are less responsive to vasoactive agents, loose their anionic charge and normal glycocalyx, become leaky and give rise to massive over-expression of nitric oxide. Disturbed gap junctions disrupt intercellular endothelial communication and thus regulation [13]. Both red and white blood cell deformability is reduced, which may cause microvascular plugging. The interaction of white blood cells and endothelium represents the crossroads between inflammation and coagulation. Numerous mediators facilitate intercellular communication and are responsible for white blood cell activation and the induction of a procoagulable state. The latter may give rise to disseminated intravascular coagulation, leading to diminished flow as a result of micro-thrombus formation. Abnormalities in the nitric oxide system induced by inflammatory activation can be regarded as one of the key mechanisms responsible for the distributive defects associated with severe sepsis and septic shock. Indeed, various studies have shown hemodynamic stabilization after blocking the inflammatory up-regulation of inducible nitric oxide synthase (iNOS) expression (for example, [5]). Inhomogeneous expression of iNOS interferes with regional blood flow and promotes shunting from vulnerable weak microcirculatory units [23]. Inhomogenous expression of endothelial adhesion molecules, such as intercellular adhesion molecules and selectines, can also be expected to contribute to distributive alterations of blood flow through its effect on white blood cell kinetics [24]. Animal experiments have shown a reduction in perfused capillary density, stopped flow next to areas of hyperdynamic blood flow, resulting in increased heterogeneity in skeletal and intestinal microvascular beds, despite frequent normo-tensive conditions [6,25]. An increased heterogeneity of the microcirculation was shown to provoke areas of hypoxia and generally impair oxygen extraction, both mathematically and in animal models of septic shock [5,25,26]. Microcirculatory PO2 measurements by palladium porphyrin phosphorescence revealed that, during various conditions of shock and resuscitation, microcirculatory PO2 levels become lower than venous PO2 levels, providing direct evidence for the action of functional shunting pathways [4,5,19,27,28]. Acidosis, hypocapnia and hypercapnia occurring during disease and therapy have been reported to have differential effects on the microcirculation, with acidosis in the presence of nitric oxide inhibition and hypocapnia causing arteriolar constriction, and hypercapnia resulting in venular dilation [29,30]. Elevated mixed venous oxygen saturation and metabolic distress, such as occurs during distributive shock, indicates a deficit in oxygen extraction rate. This may be caused by either the oxygen not reaching the microcirculation (e.g., being shunted) [27] and/or that oxygen is not being utilized by the mitochondria of the tissue cells to perform oxidative phosphorylation [31]. The latter has been termed cytopathic hypoxia [32]. This entity, combined with observed microvascular derangements, led us to introduce the term ‘microcirculatory and mitochondrial distress syndrome’ (MMDS) to identify the compartments and pathophysiology of this condition [4]. The nature of MMDS in this definition is not only defined by the condition that led to shock, the co-morbidity present and the genetic profile of the patient, but also by the length of time the condition has persisted and the treatment regime that a patient has undergone.
Classifying microcirculatory flow abnormalities in distributive shock Class Capillary hemodynamics Disease states observed in I Stagnant Pressure guided resuscitation from sepsis II Continuous/capillary fall-out On-pump CABG surgery, ECMO III Continuous/stagnant Resuscitated sepsis, reperfusion injury, sickle cell crises, malaria IV Hyperdynamic/stagnant Resuscitated sepsis V Hyperdynamic Resuscitated sepsis, exercise
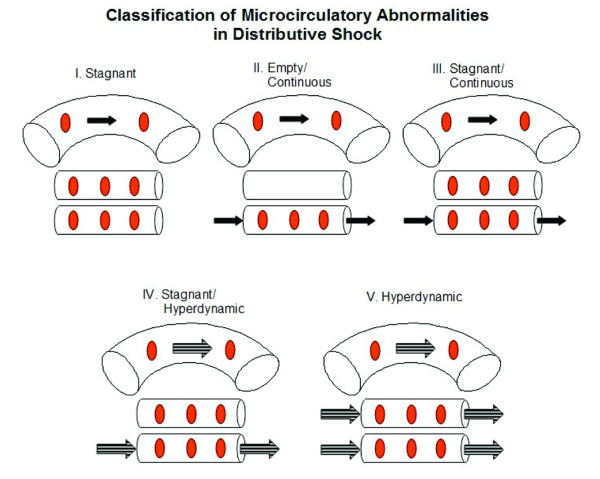
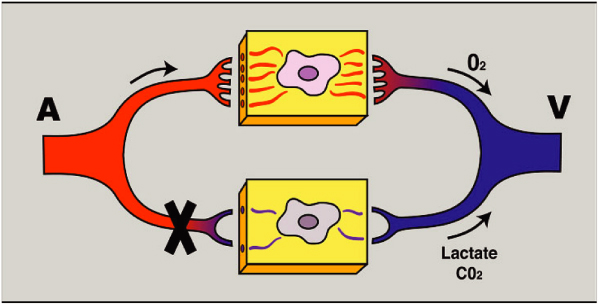
Review of microcirc in shock — ACADEMIC EMERGENCY MEDICINE 2008; 15:399413
Nitroglycerin in septic shock after intravascular volume resuscitation. Spronk PE, Ince C, Gardien MJ, Mathura KR, Oudemans-van Straaten HM, Zandstra DF. Lancet. 2002 Nov 2;360(9343):1395-6
Department of Intensive Care Medicine, Onze Lieve Vrouwe Gasthuis, Amsterdam, Netherlands. In patients with septic shock, oxygen consumption is increased, but oxygen delivery and extraction is impaired, partly because of microcirculatory shutdown and shunting. Orthogonal polarisation spectral (OPS) imaging allows visualisation of the microcirculation. We used this technique to assess microcirculatory flow in septic-shock patients who had a mean arterial blood pressure of more than 60 mm Hg and central venous pressure greater than 12 mm Hg. The infusion of 0.5 mg of nitroglycerin intravenously then resulted in a marked increase in microvascular flow on OPS imaging. Improved recruitment of the microcirculation could be a new resuscitation endpoint in septic shock.
My gut feeling is that one should open the microcirculation with a vasodilator that is independent of endothelial function like NTG, but after opening previously shut-down areas, one should probably decrease and stop NTG infusion and maintain microcirculatory perfusion with other dilators like ketanserin (it’s unfortunately more expensive, but works very well). Again, no data available and studies are difficult due to the heterogeneity of our patients. KTS seems to work especially good in the elderly atherosclerotics, while NTG may pose serious problems due to decreased vascular compliance. It makes me very humble when I realize that we go back 30-40 years, i.e. aiming for a warm toe (Shoemaker) and decreasing lactates (M.H. Weil, Science 1964).
Boosting CO with phosphodiesterase inhibitors in conjunction with (other) vasodilators like nitroglycerin and/or ketanserin works fine, especially in view of patients getting older all the time (mean age in our ICU over the last 5 years is 64). Inherent cardiac problems, i.e. trivascular impairment and especially diastolic dysfunction, are a serious problem. That’s why we don’t do “dobutamine stress tests” any more, but have been using PDE inhibitors like enoximone or milrinone instead. I know all data in this field are scarce, but these references are of interest with respect to intestinal perfusion. Schmidt W, Tinelli M, Secchi A, Gebhard M, Martin E, Schmidt H. Enoximone maintains intestinal villus blood flow during endotoxemia. Int J Surg Investig. 2001;2(5):359-67. Kern H, Schroder T, Kaulfuss M, Martin M, Kox WJ, Spies CD. Enoximone in contrast to dobutamine improves hepatosplanchnic function in fluid-optimized septic shock patients. Crit Care Med. 2001 Aug;29(8):1519-25.
Rivers Article on Microcirculatory Dysfunction (Crit Care 2005;9(Sup 4))
Microcirc Recruitment Maneuvers
(Minerva Anestesiologica 2006;72:509)
rise in tissue PCO2 via sublingual capnography or gastric tonometry may be a good marker
anaerobic CO2 production comes from the buffering of acids via bicarb
WHen hypoxia is hypoxic, CO2 levels should remain normal as blood flow is preserved
In ischemic hypoxia, CO2 rises due to stagnant blood flow
Microcirulatory Mitochondrial Distress Syndrome (MMDS)
cytopathic hypoxia
In an RCT, nitro did not result in benefit. (Crit Care Med 2010;38:93)
Terlipressin
a prodrug form of vasopressin which may be given by bolus. (Crit Care Med 2005;33(9):1897)
Digoxin
Yes, I do use digoxin, — perhaps 1 mg in three doses over 12 hours — almost routinely, in septic patients with tachycardia. I’m not sure how much it actually does control the rate and the evidence supporting its use could at best be charitably described as “low-level” (see Nasraway SA, Rackow EC, Astiz ME, Karras G, Weil MH. Inotropic response to digoxin and dopamine in patients with severe sepsis, cardiac failure, and systemic hypoperfusion. Chest. 1989 Mar;95(3):612-5.)
Levosimendan
It’s a drug that binds with troponin and acts as a calcium channel sensitiser rather than increasing intracellular concentrations of calcium. Increases cardiac output and heart rate without increasing myocardial oxygen consumption.
Levosimendan may be better than dobutamine as a inodilator (Internet J Cardiol 1528-834X)
Extravasation of Vasoconstrictors
ED Hypotension Predicts Mortality
patients with exposure to hypotension (SBP<100) without trauma
3x risk of in-house death
10x risk of sudden unexpected in-house death
any SBP <80
6x risk of in-house death
SBP<100 for >60 minutes
3x risk of in-house death
Chest 2006;130(4):941
Great Toe Temp
in one study of 100 patients the temperature of the great toe had good correlation with measured CO. (Joly HR, Weil MH. Temperature of the Great Toe as an Indication of the Severity of Shock. Circulation. 1969;Jan:131-138. prospective trial, 100 patients)
European Shock Recs
Hemodynamic monitoring in shock and implications for management International Consensus Conference (Inten Care Med 2007;33:575) 1. We recommend that shock be defined as a life-threatening, generalized maldistribution of blood flow resulting in failure to deliver and/or utilize adequate amounts of oxygen, leading to tissue dysoxia. Level 1; QoE moderate (B) 2. We recommend that hypotension [SBP < 90 mmHg, SBP decrease of 40 mmHg from baseline, or mean arterial pressure (MAP) < 65 mmHg], while commonly present, should not be required to define shock. Shock requires evidence of inadequate tissue perfusion on physical examination. Level 1; QoE moderate (B) 3. In the absence of hypotension, when shock is suggested by history and physical examination, we recommend that a marker of inadequate perfusion be measured (decreased ScvO2 , SvO2 , increased blood lactate, increased base deficit, perfusion-related low pH). Level 1; QoE moderate (B)
4. Apart from lactate and base deficit, current evidence does not support the routine use of bio-markers for diagnosis or staging of shock. Level 1; QoE high (A) 5. We recommend a target blood pressure during initial shock resuscitation of: For uncontrolled hemorrhage due to trauma: MAP of 40 mmHg until bleeding is surgically controlled. Level 1; QoE moderate (B) For traumatic brain injury (TBI) without systemic hemorrhage: MAP of 90 mmHg. Level 1; QoE low(C) For all other shock states : MAP > 65 mmHg. Level 1; QoE moderate (B) 6. We recommend that preload measurement alone not be used to predict fluid responsiveness. Level 1; QoE moderate (B) 7. We recommend that in shock, low values of commonly used static measures of preload such as CVP, RAP, PAOP (for example less than 4 mmHg) and ventricular volumes, should lead to immediate fluid resuscitation with careful monitoring. Level 1; QoE low (C) 8. We recommend a fluid challenge to predict fluid responsiveness. A fluid challenge consists of either immediate administration (for example 1015 minutes) of 250 cc of crystalloid or colloid equivalent (eventually repeatable, if indicated) or a straight-leg raise with a goal of obtaining a rise in CVP of at least 2 mmHg. A positive response includes measures of improved cardiac function and tissue perfusion. Level 1; QoE low (C) 9. We do not recommend the routine use of dynamic measures of fluid responsiveness (including but not limited to pulse pressure variation, aortic flow changes, systolic pressure variation, respiratory systolic variation test, and collapse of vena cava). Level 1; QoE high (A) There may be some advantage to these measurements in highly selected patients Level 1; QoE moderate (B) 10. We do not recommend routine measurement of CO for patients with shock. Level 1; QoE moderate (B) 11. We suggest considering echocardiography or measurement of CO for diagnosis in patients with clinical evidence of ventricular failure and persistent shock despite adequate fluid resuscitation. Level 2 (weak); QoE moderate (B) 12. We suggest serial measurements of lactates and/or base deficit as a predictor of outcome. Level 2; QoE moderate (B) 13. We do not recommend routine use of gastric tonometry, sublingual capnography, orthogonal polarization spectral (OPS) imaging and other techniques to assess regional or micro-circulation. Level 1; QoE (B) 14. a) We recommend frequent measurement of blood pressure and physical examination variables (including signs of hypoperfusion, urine output and mental status) in patients with a history and clinical findings suggestive of shock. b) We recommend invasive blood pressure measurement in refractory shock. Level 1; QoE very low (D) 15. We do not recommend the routine use of the pulmonary artery catheter for patients in shock. Level 1; QoE high (A) 16. We recommend instituting goal-directed therapy without delay, in patients presenting with septic shock (within 6 h or ideally less), particularly where ScvO 2 is below 70% Level 1; QoE moderate (B) 17. We do not recommend targeting supranormal oxygen delivery in patients with shock. Level 1; QoE high (A)
Is this patient hypovolemic?
(JAMA 1999;281:1022)
External Jugular Vein Examination
(Arch Intern Med 2006;166:2132)
Instructions given for the external jugular vein (EJV) examination (an online eVideo is available). 1, Position the patient as you would for the internal jugular vein examination, at an angle usually between 30° and 45° from the horizontal. For extremely low central venous pressure (CVP), you may need to lower this angle, and conversely you may need to seat the patient more upright to evaluate high CVP. 2, Check the left and right sides of the neck. Tangential light may help identify pulsating EJVs. If the veins are not apparent, you may ask the patient to perform a Valsalva maneuver, or place your finger at the base of the neck to distend the veins temporarily. These maneuvers will identify the course of the EJV but should not be used during measurement. 3, If the vein is readily visible and distended, locate the apex of the pulsating meniscus. (Some patients will have distended veins, but the jugular venous pressure is assessed at the top of the venous pulsation, not at the maximal height of distension. Stripping the vein will help locate the pulsations if the vein is already distended.) 4, Stripping is performed as follows: A, Place 2 adjacent fingertips over the area of interest along the EJV. B, Spread your fingers apart along the course of the EJV. C, Remove the lower finger, and leave the upper finger in place. The vein should fill from below (retrograde), and you may be able to appreciate the point of pulsation more clearly. Curved arrow indicates the head of the bed being raised upward; straight arrow, horizontal line from UC to ruler to where indicated to measure; bracket, distance on the ruler; and vertical dotted line, center of the sternum.

CVP monitored via EJ may be accurate (Anesthesiology 1973;38:291) and (Anaesthesia 2002;57:596)
Pressors do not affect Mortality
Global Perfusion and ABX, but not pressors affect mortality (Intensive Care Med. 2008 Jan;34(1):157-62.)
Differential of Low SVR
Differential of low SVR. Often considered synonymous with sepsis, but many patients had cirrhosis, pancreatitis, adrenal insufficiency (Crit Care 1999;3:71)
Methylene Blue
Article in post-liver transplant patients (Journal of Clinical Anesthesia (2012) 24, 324–328)
cardiac arrhythmias
coronary vasospasm
increased pulmonary hypertension,
MB is now recognized as a potent monoamine oxidase inhibitor.