(Liberal use of an article from Emergency Medicine Practice)
Glu to Val at pos. 6
No carbonic anhydrase inhibitors in Sicklers
Reticulocytes are early RBC that are mobilized from the BM when more RBCs are needed; Normal reticulocyte count is 0.5-1.5% Normally, reticulocytes live in bone marrow for 3 days, then spend 1 day in peripheral blood before matured. The more severe the anemia, the earlier the release; the reticulocyte may spend over 2 days in peripheral blood. Therefore, reticulocyte count should be corrected for the degree of anemia.
Corrected reticulocyte count = measured reticulocyte count X measured HCT / 45
Sickle Cell Trait
2 million people in the United States have sickle cell trait
hematuria and hyposthenuria (inability to concentrate urine)
Hemoglobin SC Disease
HbC: normal glutamic acid at position 6 of the beta chain is replaced by lysine.
Presence of HbC accentuates the deleterious effects of HbS, this makes HbSC disease a clinically significant disorder. Most complications are less frequent, less severe, and occur later in life when compared to SCA.
One exception is proliferative retinopathy, which is more common in those with HbSC disease.
Exam
Ask about pneumovax
Cardiac murmurs are frequent in patients with SCD and do not necessarily indicate acute pathology. In one series of 100 patients with SCD, nearly 80% had murmurs.
Ask if this event is different from previous crises
CBC, Reticulocyte Count, T+H
Get a routine UA, low threshold for C-XR
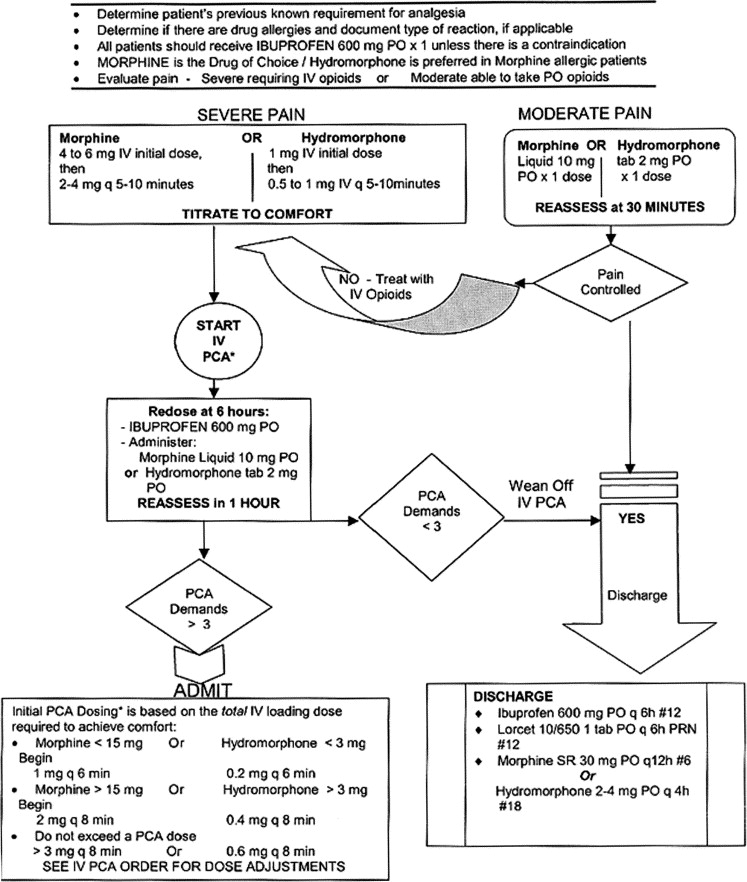
Initial Treatment
Aggressive IV hydration may enhance the risk for the development of non-cardiogenic pulmonary edema and/or acute chest syndrome in a patient with vaso-occlusive crisis.
Vaso-occlusive (Thrombotic) Crisis
Severe pain in the back, abdomen, chest, and/or long bones. Search for infectious source.
Admission Criteria for Vaso-Occlusive Crisis
Inability to control pain in ED
Profound or persistent tachycardia
Hypotension
Temperature > 101°F
Significant infection
Aplastic or hyperhemolytic crisis (acute fall of hemoglobin > 1 g/dL)
Abnormal chest x-ray
Prolonged priapism
New CNS findings
Acute abdomen
Significant hypoxia or acidosis
Pregnancy
Hepatic syndrome or cholecystitis
Ischemia to viscosity to sludging to obstruction
 (J EM 2007;April)
(J EM 2007;April)
Hyperhemolytic Crisis
Hyperhemolytic crisis (HC) involves a higher-than-normal rate of hemolysis and often occurs in conjunction with a vaso-occlusive crisis.
It is occasionally precipitated by infection
Hyperhemolysis can also occur when an individual with SCA has a co-existent G6PD deficiency and is given certain medications, such as sulfa drugs or nitrofurantoin.
In addition to pain, patients with HC may present with fatigue, increased scleral icterus, and jaundice.
On laboratory analysis, HC is characterized by a decrease in hemoglobin, higher-than-usual reticulocyte count, increased indirect bilirubin, and increased LDH. HCs are typically self-limited, but transfusion may be necessary for severe anemia.
Acute Splenic Sequestration
spleen suddenly traps a large number of red cells causing severe anemia (decrease in hemoglobin by 20% or more), an enlarging spleen (by at least 2 cm from baseline), hypovolemia, and mild thrombocytopenia.
Incidence peaks at 1-2 years of age and is most common in children with HbS disease.
It can recur, up to 50% of children will have a second episode, usually within two years.
the presentation is usually striking, children will usually arrive in extremispale, tachycardic, and hypotensive. A massive spleen will dominate the belly. Diagnostic tests apart from a stat hemoglobin are generally unnecessary, but a bedside ultrasound of the abdomen could help rule out intraperitoneal fluid (if splenic rupture is suspected). Once the child is stabilized, CT scan of the abdomen may be helpful if the diagnosis remains in doubt or if there is concern for a splenic abscess.
Rx:
Hypovolemic shock is more lethal than anemia, immediately give large amounts of IV crystalloid (20-40 cc/kg)
After stabilization of blood pressure, begin the transfusion.
After transfusion, hemoglobin can then rise dramatically when spleen releases RBCs, and the increased blood viscosity will worsen perfusion. For these reasons, some experts suggest utilizing an exchange transfusion to avoid this complication in seriously ill patients with acute splenic sequestration
goal of transfusion during acute splenic sequestration is to achieve a post-transfusion hemoglobin level of 6-8 g/dL.
Patients may need full or partial splenectomy
Aplastic Crisis
Often precipitated by infection. usually Human Parvovirus B19 which suppresses erythropoiesis.
The patient will be pale, tachycardic, and fatigued.
On labs decreased reticulocyte count will be seen.
Treat with blood transfusions, fluids, and treatment of any infection.
During an aplastic crisis, there is a temporary arrest of red cell production; thus, reticulocytopenia (generally < 2%) is the hallmark of this condition. Patients will demonstrate a variable decrease in hemoglobin. Usually only erythropoiesis is affected, but neutropenia and thrombocytopenia are occasionally seen.6 Aplastic crisis is often caused by parvovirus B19. In one study, 80% of such cases of aplastic crisis were associated with this organism.119
Aplastic crisis most commonly occurs in children and usually resolves spontaneously within 5-10 days. Treatment is mainly supportive, but transfusion may be necessary. Isolation is warranted to prevent contact with pregnant women and other sickle cell patients. Indications for transfusion include 25% or greater decrease in hemoglobin level from baseline with a low reticulocyte count and severe symptoms from the anemia.120
Acute Chest Syndrome (ACS)
New pulmonary infiltrate, chest pain, fever, and hypoxia/the appearance of a new infiltrate on chest x-ray accompanied by acute respiratory symptoms
CAP agents are the main cause in addition to contribution from thrombosis and thromboembolism.
2 of the following: chest pain, fever, infiltrate or V/Q abnormality, respiratory symptoms, hypoxemia
Adult patients with ACS tend to be afebrile, have severe pain, and often have multilobar disease combined with high mortality.
In contrast, children are more often febrile and usually present with cough without chest pain.
While the chest film is important in diagnosing ACS, the initial radiograph is normal in almost half of patients who ultimately develop the syndrome. Even when positive, chest radiography often underestimates vascular damage and accompanying physiologic derangement. The degree of hypoxia measured by pulse oximetry or by arterial blood gas is usually out of proportion to the findings seen on chest x-ray.
Rx
Early administration of broad-spectrum antibiotics
One randomized, double-blind, placebo-controlled trial showed that IV dexamethasone (0.3 mg/kg q12h x 4 doses) had a beneficial effect in hospitalized children with mild to moderately severe ACS. Mean hospital stay was shorter in the dexamethasone-treated group, and the steroids prevented clinical deterioration and reduced the need for blood transfusions.
Some patients with ACS may require exchange transfusions.
Always consider PE in the differential though it is nearly impossible to distinguish between ACS and PE in the ED.
Infectious Sequelae
Immune defect for encapsulated organisms, as sicklers are often functionally asplenic
Encapsulated organisms such as Streptococcus pneumoniae and Escherichia coli, Klebsiella sp., and Salmonella sp.
Absolute band count of >3000. Children with high white counts should always be cultured and treated with broad spectrum ABX.
Bone and Joint Pain
Differential is crisis pain vs. bone infarction vs. osteomyelitis (often secondary to salmonella)
Infarction and infection usually present with fever and wbc above baseline
Joint Pain
Osteonecrosis is a painful and often disabling complication of repeated bony infarctions. By age 35, half of all sickle cell patients have evidence of hip and shoulder osteonecrosis.6 Patients complain of pain and/or limited range of motion of the affected joint. In the early stages of disease, radiographs will often appear normal,29 although MRI may be diagnostic.6
The most important entity to rule out in a sickle cell patient with joint pain is septic arthritis. Suspect septic arthritis when joint pain is accompanied by fever, significant pain on range of motion, or erythema/edema involving the joint. Joint aspiration is the most vital (and the only definitive test) in such cases.
Dactylitis
Also known as hand-foot syndrome, dactylitis may be the earliest manifestation of SCD.48,112 This condition is usually seen in children younger than 6 months of age, but can occur up to 4 years of age.19,112 Physical exam reveals swelling of the hands or feet, pain, and fever. There may also be erythema, mimicking osteomyelitis,113 and the WBC count and sedimentation rate may be elevated. Initial radiographs are normal, but radiographs taken several days after the onset of symptoms may show periosteal elevation. Treatment is supportive, with analgesia, hydration, and warm compresses. Symptoms are usually self-limited.80
Abdominal Pain
Potential Causes Of Abdominal Pain In Sickle Cell Anemia
- Spleen
- Sequestration
- Hemorrhage
- Infarction
- Abscess
- Hepatobiliary
- Hepatitis
- Cholelithiasis/cholecystitis
- Hepatic sequestration
- Intrahepatic cholestasis
- Renal
- Stone
- Clot
- Papillary necrosis
- Cystitis
- Bone
- Infarction: ribs, spine, femoral head
- Osteonecrosis
- Vertebral collapse
- Miscellaneous
- Pneumonia
- Mesenteric ischemia
One study retrospectively examined findings in 53 patients with SCA who presented with abdominal pain.30 A vaso-occlusive crisis was responsible for the pain in 57%, while 23% had a surgical entity and 20% had a nonsurgical genitourinary disorder. Overall, 77% of the patients with painful sickle crisesbut no patient with an acute surgical processcomplained of coexistent abdominal and remote (usually extremity) pain. In this series, all patients with a surgical condition complained of localized rather than diffuse pain. Of note, laboratory parameters, including the leukocyte count, did not distinguish sickle crisis from a surgical condition. (Am Surg 1989;55(11))
Biliary tract and parenchymal liver disease are the most prevalent and serious complications that affect the digestive system.6 Five hepatobiliary syndromes that are especially common include viral hepatitis, hepatic crisis, cirrhosis, cholelithiasis with or without cholecystitis, and intrahepatic cholestasis.31 Of these, the most concerning diagnosis is intrahepatic cholestasis. It is characterized by sudden onset of severe right upper quadrant pain, progressive hepatomegaly, coagulopathy, and extreme hyperbilirubinemia.28,31 Treatment is supportive, with transfusion and correction of coagulopathy. The mortality is very high.
Genitourinary Complications
There are a number of genitourinary complications seen in SCA. The primary renal complications seen in SCD include hematuria, nephrotic syndrome, and, in rare circumstances, renal failure.
Hematuria is usually self-limited, resolving with bed rest alone. It may occur in those with sickle cell trait, and this diagnosis should be considered in the patient of African descent who presents with painless hematuria.137 Transfusion may be needed if the hematuria is severe.138 Years of glomerular hyperfiltration results in renal damage and the inability to concentrate urine (isothenuria), but chronic renal failure in SCD is uncommon overall.6 A prospective longitudinal study followed a cohort of 725 patients with SCA and found chronic renal failure in only 4.2 %.139
Priapism
Occurs in 10-40% of male Sicklers and can result in permanent damage.
Urologic consult and .25-.5 mg of SQ terbutaline (not in penis)
Hydrate, transfuse, and possibly hyperbaric O2.
Priapism occurs when sickled cells congest the corpora and prevent emptying of blood from the penis. It is a prolonged, usually painful penile erection not initiated by sexual stimuli.140 The priapism that is seen in sickle cell patients is termed a “low flow” or ischemic priapism. Low-flow priapism is a time-sensitive emergency, as irreversible cellular damage and fibrosis can occur if treatment is not administered in 24-48 hours.140 (A full discussion on priapism can be found in the November 2000 issue of Emergency Medicine Practice, “Male Genitourinary Emergencies: Preserving Fertility And Providing Relief.”) Treatment of priapism in sickle cell patients includes analgesia, hydration, oxygen, and, occasionally, exchange transfusion. These measures are successful in about 80% of patients.141 If simple interventions fail, corporal aspiration and injection of a vasoconstrictor are occasionally successful in sickle cell patients.141 Urology consultation is indicated.
intracavernosal epinephrine 1:1,000,000
Ophthalmologic Complications
The most serious ocular emergency in sickle cell patients is hyphema in the setting of trauma. The sickled cells tend to obstruct the flow of aqueous humor and may result in elevation of intraocular pressure and subsequent acute angle closure glaucoma. If the intraocular pressure is 24 mmHg or greater in the setting of hyphema, treatment for acute angle closure glaucoma should be initiated. Urgent ophthalmologic consultation should be obtained in all sickle cell patients with hyphema.115
Give atropine drops but do not give diamox/acetazolamide as it precipitates sickling
labs
send cbc, lfts, and retic counts in almost all cases
be gentle with hydration; excess fluids have been linked to atelectasis which can lead to ACS
Use D5 1/2 NS per the EM practice article,
which actually makes no sense, b/c acidosis promotes sickling
D5LR is probably a better choice
no supplemental O2