THE FOUR MOST IMPORTANT EQUATIONs
By Lawrence Martin, M.D., FACP, FCCP
Associate Professor of Medicine
Case Western Reserve University School of Medicine
Cleveland, Ohio
Disclaimer Contents: Summary The PCO2 Equation The Henderson-Hasselbalch Equation Alveolar Gas Equation Oxygen Content Equation References True – False Quiz Abbreviations used: CaO2 – arterial oxygen content CO – carbon monoxide COHb-carboxyhemoglobin DKA– diabetic ketoacidosis FIO2
– fraction of inspired oxygen Hb
– hemoglobin HCO3– bicarbonate H-H – Henderson-Hasselbalch ICU
– Intensive care unit metHb– methemoglobin PaCO2 – arterial PCO2 in mm Hg PACO2– alveolar PCO2 in mm Hg PaO2– arterial PO2 in mm Hg PAO2– alveolar PO2 in mm Hg PB– barometric pressure in mm Hg R– respiratory quotient SaO2– % saturation of hemoglobin with O2 VA– alveolar ventilation in L/min VCO2– CO2 production in ml/min VD– dead space ventilation VE– minute or total ventilation V-Q – ventilation-perfusion
Four equations are taught briefly in medical school but are grossly under-emphasized in importance and are therefore invariably forgotten in later years, when they are most needed. The reasons why these highly important equations are ‘under taught’ in medical school are several:
- a crowded curriculum that must make room for immunology and cell biology
- the teachers may have little or no clinical experience with respiratory patients, and therefore can’t possibly know how important these equations are in the everyday practice of medicine
- misguided leadership of curriculum committees that may feel every subject deserves equal balance, and thus leave it up to the student to ‘learn it all’ without anyone guiding them as to what is really important in the care of patients. (For example, one hour on surfactant may be equally weighted with one hour on gas exchange, which may be OK for training Ph.D.’s but is misguided for training physicians).
These four equations express relationships that are extremely important in clinical practice. They are the:
- PCO2 equation
- Henderson-Hasselbalch equation
- Alveolar Gas equation, and
- Oxygen Content equation.
Emphasis should be placed on understanding the simple qualitative relationships expressed by these equations. Each equation can be clinically applied in the assessment of abnormal oxygenation, ventilation, or acid-base balance. For example, variables in the PCO2 equation, and not any bedside observations, define the common terms hyperventilation and hypoventilation and explain why a dyspneic, tachypneic patient may be retaining CO2. Ignorance of this and other relationships expressed in the four equations is reflected in some common diagnostic and therapeutic mistakes.
INTRODUCTION
There is disparity between the physiology we teach and expect medical students to learn and the physiology that medical residents and practicing physicians seem to know and understand. This disparity is perhaps best exemplified by four simple equations important in understanding cardiopulmonary and renal disorders (Table I). These equations are seldom emphasized beyond medical school, yet not appreciating the physiology behind them can (and often does) lead to clinical errors.
Intensive care units have contributed to the weakening knowledge of physiology among primary care physicians. Today, the more profound physiologic derangements are usually managed in ICUs by organ-specific specialists; these derangements (e.g., shock, pulmonary edema, acute ventilatory failure, acute renal failure) are literally outside the care of most physicians and surgeons. Not all serious physiologic problems are handled in ICUs however, and the need for understanding basic physiology – in the office, on the general medical wards – remains paramount.
The four equations in this paper (Table I, below) are important clinically not so much for the numbers they generate as for their qualitative relationships. All four equations can be abbreviated to simpler terms that are adequate for most clinical purposes.
TABLE I: THE FOUR MOST IMPORTANT EQUATIONS
IN CLINICAL PRACTICE Equation Title Complete Equation Abbreviation Sufficient for
Most Clinical Applications
1. PCO 2 equation
PACO2=VCO2 x 0.863 / VA
where VA=VE-VD PaCO2 ~ VCO2 / VA
2. Henderson-Hasselbalch equation
pH=pK + log HCO3- / 0.03(PaCO2) pH ~ HCO3- / PaCO2
3. Alveolar gas equation
PAO2=FIO2(PB-PH2o)-PACO2[FIO2 + (1-FIO2) / R] PAO2=FIO2(PB-47)-1.2(PaCO2)
4. Oxygen content equation
CaO2=(SaO2 x Hb x 1.34) + .003(PaO2)
where:
1.34=ml O2/gram Hb
.003=ml O2/mm Hg PaO2/dl
Hb=content in grams/dl CaO2=SaO2 x 1.34 x Hb
Obviously other equations besides those in Table 1 can be important in assessing disordered physiology. The point is not to belabor equations but to emphasize a few key relationships often overlooked or misapplied in the daily practice of medicine. By understanding them we can take better care of our sickest patients wherever they are encountered. Non-intensivists should be familiar with these particular equations and their clinical relevance. They were probably all learned by most physicians at one time. For physicians in training and practitioners alike, now is the time to review. 1. The PCO2 Equation Top
The PCO2 equation puts into physiologic perspective one of the most common of all clinical observations: a patient’s respiratory rate and breathing effort. The equation states that alveolar PCO2 (PACO2) is directly proportional to the amount of CO2 produced by metabolism and delivered to the lungs (VCO2) and inversely proportional to the alveolar ventilation (VA). While the derivation of the equation is for alveolar PCO2, its great clinical utility stems from the fact that alveolar and arterial PCO2 can be assumed to be equal. Thus:
VCO2 x 0.863 PaCO2 = —————— VA The constant 0.863 is necessary to equate dissimilar units for VCO2 (ml/min) and VA (L/min) to PACO2 pressure units (mm Hg). Alveolar ventilation is the total amount of air breathed per minute (VE; minute ventilation) minus that air which goes to dead space per minute (VD). Dead space includes all airways larger than alveoli plus air entering alveoli in excess of that which can take part in gas exchange. Even when alveolar and arterial PCO2 are not equal (as in states of severe ventilation-perfusion imbalance), the relationship expressed by the equation remains valid: VCO2 PaCO2 = —————— VA In the clinical setting we don’t need to know the actual amount of CO2 production or alveolar ventilation. We just need to know if VA is adequate for VCO2; if it is, then PaCO2 will be in the normal range (35-45 mm Hg). Conversely, a normal PaCO2 means only that alveolar ventilation is adequate for the patient’s level of CO2 production at the moment PaCO2 was measured. From the PCO2 equation it is evident that a level of alveolar ventilation inadequate for CO2 production will result in an elevated PaCO2 (> 45 mm Hg; hypercapnia). Thus patients with hypercapnia are hypoventilating (the term hypoalveolarventilating would be more appropriate but hypoventilating is the conventional term). Conversely, alveolar ventilation in excess of that needed for CO2 production will result in a low PaCO2 (< 35 mm Hg; hypocapnia) and the patient will be hyperventilating. (Confusion sometimes arises because the prefix (hyper-, hypo-) differs for the same condition depending on whether one is describing a blood value or the state of alveolar ventilation.) For reasons that will be discussed below, the terms hypo- and hyper- ventilation refer onlyto high or low PaCO2, respectively, and should not be used to characterize any patient’s respiratory rate, depth, or breathing effort.
From the PCO2 equation it follows that the only physiologic reason for elevated PaCO2 is a level of alveolar ventilation inadequate for the amount of CO2 produced and delivered to the lungs.1 Thus arterial hypercapnia can always be explained by:=”http:>
- not enough total ventilation (as may occur from central nervous system depression or respiratory muscle weakness); or
- too much of the total ventilation ending up as dead space ventilation (as may occur in severe chronic obstructive pulmonary disease, or from rapid, shallow breathing); or
- some combination of 1) and 2).
Excess CO2 production is omitted as a specific cause of hypercapnia because it is never a problem for the normal respiratory system unimpeded by a resistive load. During submaximal exercise, for example, where CO2 production is increased, PaCO2 stays in the normal range because VA rises proportional to the rise in VCO2. With extremes of exercise (beyond anaerobic threshold) PaCO2 falls as compensation for the developing lactic acidosis.2 In health PaCO2 may be reduced but is never elevated.
An important clinical corollary of the PaCO2 equation is that we cannot reliably assess the adequacy of alveolar ventilation – and hence PaCO2 – at the bedside. Although VE can be easily measured with a handheld spirometer (as tidal volume times respiratory rate), there is no way to know the amount of VE going to dead space or the patient’s rate of CO2 production. A common mistake is to assume that because a patient is breathing fast, hard and/or deep he or she must be “hyperventilating.” Not so, of course.
CASE 1 A house officer was called to the bedside of an elderly woman patient late at night. She was in hospital for evaluation of a pelvic mass. The patient was noted to be anxious and complaining of shortness of breath; her lung fields were clear to auscultation and vital signs were normal except for slight tachycardia and respiratory rate of 30/minute. A nurse commented that the patient “gets like this every night.” The physician ordered a benzodiazepine drug for what he described as “hyperventilation and anxiety.” Thirty minutes later the patient’s breathing slowed considerably and she became cyanotic, whereupon she was transferred to the ICU.
Although nothing in the PCO2 equation directly relates respiratory rate or depth of breathing to PaCO2, physicians commonly (and mistakenly) use these observations to assess a patient’s PaCO2. The error in this case was to assume the patient was hyperventilating (because she was breathing fast) and could tolerate the sedative; in fact she was hypoventilating – her PaCO2 was elevated (as will be explained further under Equation 2).
Hypercapnia represents a failure of the respiratory system in some aspect and therefore a state of severe organ system impairment. In addition to this clinical fact there are three physiologic reasons why elevated PaCO2 is potentially dangerous. First, as PaCO2 increases, unless HCO3- also increases by the same degree pH will fall (see Equation 2). Second, as PaCO2 increases PAO2 (and hence PaO2) will fall unless inspired oxygen is supplemented (see Equation 3). Third, the higher the PaCO2, the less defended is the patient against any further decline in alveolar ventilation.
This last point is graphically illustrated by plotting PaCO2 against alveolar ventilation Figure 1. The higher the PaCO2 is to begin with, the more it will rise for any given decrement in alveolar ventilation. For example a decrease in alveolar ventilation of one L/minute (as may occur from anesthesia, sedation, congestive heart failure, etc.) will increase a baseline PaCO2 of 30 mm Hg to 36.3 mm Hg when VCO2 is 200 ml/min; the same one L/min decline in VA will raise a baseline PCO2 of 60 mm Hg to 92 mm Hg Figure 1). Whereas the hyperventilating or normally- ventilating patient can almost always tolerate sedating drugs (without clinically important hypoventilation), even a small amount of sedative may be dangerous in the hypercapnic patient.
Note also from Figure 1 that an increase in CO2 production (e.g., from 200 to 300 ml/min) without concomitant increase in VA (as should occur normally) will cause PaCO2 to increase. This situation is sometimes seen in patients with severe chronic obstructive lung disease when they exercise, and in artificially-ventilated patients who are carbohydrate loaded (which increases CO2 production). The basic mechanism for hypercapnia in these and all other cases, however, is inadequate VA for the amount of CO2 delivered to the lungs.
 2. The Henderson-Hasselbalch Equation Top
2. The Henderson-Hasselbalch Equation Top
Of the four equations in this paper, the Henderson-Hasselbalch is the one with which physicians are most familiar. The H-H equation is repeatedly emphasized in basic science courses and in renal and pulmonary pathophysiology lectures; students hear about it on many occasions.
The bicarbonate buffer system, quantitatively the largest in the extracellular fluid, instantaneously reflects any blood acid-base disturbance in one or both of its buffer components (HCO3- and PACO2). The ratio of HCO3- to PACO2 determines pH and therefore the acidity of the blood:
HCO3- pH = pK + log ———————- 0.03 (PaCO2) pH is the negative logarithm of the hydrogen ion concentration, [H+], in nM/L (nM = nanomole = 1 x 10-9 moles; pH 7.40 = 40 nM/L [H+]). Because of the negative logarithm, small numerical changes of pH in one direction represent large changes of [H+] in the other direction (Table II). An 0.1 unit fall in pH from 7.4 to 7.3 represents a 25% increase in [H+]; a similar percentage change in serum sodium would increase its value from a normal 140 mEq/L to 175 mEq/L! TABLE II: pH and Hydrogen Ion Concentration Top Blood pH [H+] (nM/L) % Change from normal Acidemia 7.00 100 + 150 7.10 80 + 100 7.30 50 + 25 Normal 7.40 40
Alkalemia7.52 30 – 25 7.70 20 – 50 8.00 10 – 75
Unfortunately, the logarithmic nature of pH and the fact that acid-base disorders involve simultaneous changes in three biochemical variables and in the function of two organ systems (renal and respiratory), have all combined to made acid-base a difficult subject for many clinicians. In the 1970s nomograms incorporating the H-H variables and compensation bands for the four primary acid-base disorders were introduced as aids to determining a patient’s acid-base status.3-8 While nomograms can be helpful if readily available and properly used, there is much to be gained by simply knowing the relationship among the three H-H variables and the type of changes expected with each disorder. In this regard the following items of clinical importance bear emphasis.
a) If any of the three H-H variables is truly abnormal the patient has an acid-base disturbance without exception. Thus any patient with an abnormal HCO3- or PaCO2, not just abnormal pH, has an acid-base disorder. Most hospitalized patients have at least one bicarbonate measurement as part of routine serum electrolytes; this is usually called the ‘CO2’ or ‘total CO2’ when measured in venous blood. (Total CO2 includes bicarbonate and the CO2 contributed by dissolved carbon dioxide, the latter 1.2 mEq/L when PaCO2 is 40 mm Hg. For this reason, and because bicarbonate concentration is slightly higher in venous than in arterial blood, total CO2 runs a few mEq/L higher than the bicarbonate value calculated using the H-H equation.) If total CO2 is truly abnormal the patient has an acid-base disorder. In Case 1 there were two sets of electrolyte measurements on the patient’s chart when the sedative was ordered; both showed total CO2 elevated at 34 mEq/L. The patient had been taking a diuretic so it was probably assumed that her elevated total CO2 reflected a mild metabolic alkalosis. More likely, however, it represented chronic respiratory acidosis with renal compensation. When she arrived to the ICU her arterial blood gas showed pH 7.07, PaCO2 83 mm Hg, PaO2 55 mm Hg (breathing supplemental oxygen), HCO3- 23 mEq/L, values that reflected a worsening of previously- unrecognized respiratory acidosis plus a new metabolic acidosis (lactic acidosis from decreased organ perfusion). The patient’s long smoking history and the physical findings suggested chronic obstructive lung disease (later confirmed by pulmonary function tests). Her anxiety prior to MICU transfer was related to worsening acidosis and dyspnea.
b) The simplified version of the H-H equation eliminates the log and the pK, and expresses the relationships among the three key values.
HCO3- pH ~ ———————- PaCO2 This version is sufficient for describing the four primary acid-base disturbances and their compensatory changes listed in Table III (below). If the numerator is first to change the problem is either metabolic acidosis (reduced HCO3-) or metabolic alkalosis (elevated HCO3-); if the denominator is first to change the problem is either respiratory alkalosis (reduced PaCO2) or respiratory acidosis (elevated PaCO2).
TABLE III. The four primary acid-base disorders and their compensatory changes. The primary event leads to a large change in pH (larger arrows). Compensation (changes in HCO3- and PaCO2 represented by smaller arrows) attempts to normalize the ratio of HCO3-/PaCO2 and bring the pH back toward normal (smaller arrows next to pH). Each primary disorder may be caused by a variety of specific clinical conditions (see text).
Top
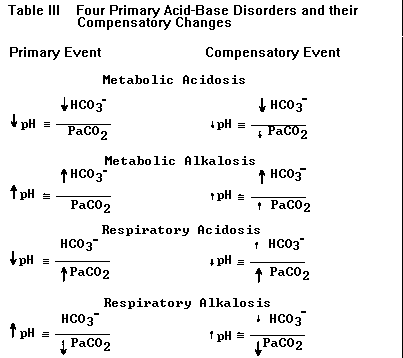 c) By convention ‘acidosis’ and ‘alkalosis’ refer to in-vivo physiologic derangements and not to any change in pH. Each primary acid-base disorder arises from one or more specific clinical conditions, e.g., metabolic acidosis from diabetic ketoacidosis or hypoperfusion lactic acidosis; metabolic alkalosis from diuretics or nasogastric suctioning; etc. Thus the diagnosis of any primary acid-base disorder is analogous to diagnoses like “anemia” or “fever”; a specific cause must be sought in order to provide proper treatment. Because of the presence of more than one acid-base disorder (‘mixed disorders’) a patient with any acidosis or alkalosis may end up with a high, low or normal pH. For example, a patient with obvious metabolic acidosis from uremia could present with a high pH due to a concomitant metabolic alkalosis (which may not be as clinically obvious). Acidemia (low pH) and alkalemia (high pH) are terms reserved for derangements in blood pH only.
c) By convention ‘acidosis’ and ‘alkalosis’ refer to in-vivo physiologic derangements and not to any change in pH. Each primary acid-base disorder arises from one or more specific clinical conditions, e.g., metabolic acidosis from diabetic ketoacidosis or hypoperfusion lactic acidosis; metabolic alkalosis from diuretics or nasogastric suctioning; etc. Thus the diagnosis of any primary acid-base disorder is analogous to diagnoses like “anemia” or “fever”; a specific cause must be sought in order to provide proper treatment. Because of the presence of more than one acid-base disorder (‘mixed disorders’) a patient with any acidosis or alkalosis may end up with a high, low or normal pH. For example, a patient with obvious metabolic acidosis from uremia could present with a high pH due to a concomitant metabolic alkalosis (which may not be as clinically obvious). Acidemia (low pH) and alkalemia (high pH) are terms reserved for derangements in blood pH only.
CASE 2. A 31-year-old woman presented to the emergency room with mild diabetic ketoacidosis (DKA) and dyspnea; arterial pH was 7.25, PaCO2 34 mm Hg, HCO3- 16 mEq/L, blood sugar 475 mgm%. Her breathing difficulty was attributed to Kussmaul-type respirations characteristic of DKA. Judging her DKA non-critical, the admitting physician placed her on a general medical ward and began appropriate treatment with insulin and fluids. Four hours later she appeared more dyspneic; repeat blood gas showed pH 7.18, PaCO2 49 mm Hg, HCO3- 18, blood sugar 350 mgm%. She was transferred to MICU where she was noted to be wheezing; bronchodilator therapy was begun. Her pre-bronchodilator peak expiratory flow rate was 110 L/min, 25% of predicted. Two days later her ketoacidosis was fully corrected and peak flow was recorded at 350 L/min.
g) Acute, uncompensated respiratory alkalosis (acute hyperventilation) and acidosis (acute hypoventilation) cause predictable changes in pH and bicarbonate11,12 (Table IV). Bicarbonate increases slightly from the biochemical reaction of acutely retained CO2 and decreases when CO2 is acutely excreted;11,12 these changes are instantaneous and independent of any renal compensation. Extreme acute hyperventilation can lower the bicarbonate to about 15 mEq/L and extreme acute hypoventilation can raise it to about 29 mEq/L (Table IV); a bicarbonate value outside this range must indicate either a renal compensatory mechanism or a primary metabolic acid-base disorder. The biochemical changes in bicarbonate from acute shifts in PaCO2 point to another particularly useful clue to the presence of a mixed disorder: a higher- or lower-than- expected bicarbonate value with any change in PaCO2. Thus a slightly low HCO3- concentration in the presence of hypercapnia suggests a concomitant metabolic acidosis (e.g., PCO2 50 mm Hg, pH 7.27, HCO3- 22 mEq/L); a slightly elevated HCO3- in the presence of hypocapnia suggests a concomitant metabolic alkalosis (e.g. PCO2 30 mm Hg, pH 7.56, HCO3- 26 mEq/L).
TABLE IV. Changes in arterial pH and bicarbonate with acute changes in PaCO2. The ranges represent the 95% confidence limits for pH and bicarbonate when PaCO2 changes acutely (before any renal compensation takes place). Note that bicarbonate decreases with acute hyperventilation and increases with acute hypoventilation. (Data from references 11-12).
TABLE IV
Top PaCO2 (mm Hg) pH HCO3-15 7.61-7.74 15.3-20.5 20 7.55-7.66 17.7-22.8 30 7.45-7.53 21.0-25.6 40 7.38-7.45 22.8-26.8 50 7.31-7.36 24.1-27.5 60 7.24-7.29 25.1-27.9 70 7.19-7.23 25.7-28.5 80 7.14-7.18 26.2-28.9 90 7.13-7.09 26.5-29.2
h) The bicarbonate (or total CO2) should also be examined in relation to the other measured electrolytes, specifically to calculate the anion gap (AG). AG is the Na+ concentration minus (total CO2 + Cl-). The normal AG, 12 +/- 4 mEq/L, is an artifact of measurement since these three electrolytes are only the ones most commonly measured. (Since the value of K+ is small and relatively constant it is not usually used to calculate the AG; if K+ is used then the normal AG is about 16 +/- 4 mEq/L). If all the serum anions and cations were measured anions would equal cations and there would be no anion gap. The importance of the anion gap is that it can help both to diagnose the presence of a metabolic acidosis and characterize its cause. Thus, regardless of pH an elevated AG suggests a metabolic acidosis from unmeasured organic anions, e.g., lactic acidosis or ketoacidosis;19-21 the higher the AG the more likely it reflects an organic acidosis.19 On the other hand a normal AG in a patient with metabolic acidosis indicates a hyperchloremic acidosis, most commonly from renal or gastrointestinal bicarbonate loss, e.g., renal tubular acidosis or diarrhea.
The alveolar gas equation for calculating PAO2 is essential to understanding any PaO2 value and in assessing if the lungs are properly transferring oxygen into the blood. Is a PaO2 of 28 mm Hg abnormal? How about 55 mm Hg? 95 mm Hg? To clinically interpret PaO2 one has to also know the patient’s PaCO2, FIO2 (fraction of inspired oxygen) and the PB (barometric pressure), all components of the equation for PAO2:
<>1-FIO2 <>PAO2 = FIO2(PB-PH20) – PACO2 <>[ FIO2 + —————–] <>R
Despite this undisputed physiologic fact physicians sometimes make clinical decisions
based on PaO 2 alone, without reference to the calculated PAO2.
The abbreviated equation below is useful for clinical purposes; in this version alveolar PO2 equals inspired PO2 (PIO2) minus arterial PCO2 x 1.2, assuming the R value is 0.8 (and assuming identical values for arterial and alveolar PCO2. Water vapor pressure in the airways is dependent only on body temperature and is 47 mm Hg at normal body temperature (37 degrees C).
PAO2 = FIO2(PB-47) – 1.2(PaCO2)
Ambient FIO2 is the same at all altitudes, 0.21. It is usually not necessary to measure PB if you know its approximate average value where the blood was drawn (e.g. sea level 760 mm Hg; Cleveland 747 mm Hg; Denver 640 mm Hg). In the abbreviated equation PaCO2 is multiplied by 1.2, a factor based on assumed respiratory quotient (CO2 excretion over O2 uptake in the lungs) of 0.8; this factor becomes 1.0 when the FIO2 is 1.0.22 The following comments are meant to show how the alveolar gas equation can be clinically helpful without the need for anything more than mental calculation.
a) If PIO2 is held constant and PaCO2 increases, PAO2 and PaO2 will always decrease. Since PAO2 is a calculation based on known (or assumed) factors, its change is predictable. PaO2, by contrast, is a measurement whose theoretical maximum value is defined by PAO2 but whose lower limit is determined by ventilation-perfusion (V-Q) imbalance, pulmonary diffusing capacity and oxygen content of blood entering the pulmonary artery (mixed venous blood). In particular, the greater the imbalance of ventilation-perfusion ratios the more PaO2 tends to differ from the calculated PAO2. (The difference between PAO2 and PaO2 is commonly referred to as the ‘A-a gradient.’ However, ‘gradient’ is a misnomer since the difference is not due to any diffusion gradient, but instead to V-Q imbalance and/or right to left shunting of blood past ventilating alveoli. Hence ‘A- a O2 difference’ is the more appropriate term.)
b) The alveolar-arterial PO2 difference, notated P(A-a)O2, varies normally with age and FIO2. Up to middle age, breathing ambient air, normal P(A-a)O2 ranges between 5 and 20 mm Hg. Breathing an FIO2 of 1.0 the normal P(A-a)O2 ranges up to about 110 mm Hg23. If P(A-a)O2 is increased above normal there is a defect of gas transfer within the lungs; this defect is almost always due to V-Q imbalance.
CASE 3 A 27-year-old young woman came to the emergency room complaining of pleuritic chest pain of several hours duration. She was not a smoker but gave a history of using birth control pills. Her chest x-ray and physical exam were normal except for splinting with deep inspirations. Arterial blood gas showed pH 7.45, PaCO2 31 mm Hg, HCO3- 21 mEq/L, PaO2 83 mm Hg (breathing ambient air; PB 747 mm Hg). She was presumptively diagnosed as having pleurodynia and discharged with pain medication.
This young woman’s PaO2 was initially judged ‘normal’ and so an abnormality in oxygen transfer was missed. The calculated PIO2 and PAO2 were 147 mm Hg and 110 mm Hg, respectively. Her P(A-a)O2 was elevated at 27 mm Hg (110 minus 83), indicating a state of V-Q imbalance, and therefore some parenchymal lung disease or abnormality. Indeed, she returned the next day with similar complaints, at which time a lung scan showed defects interpreted as high probability for pulmonary embolism.
c) Because of several assumptions in clinical use of the alveolar gas equation, precision in calculating PAO2 is not achievable.22 Fortunately an estimate of P(A-a) O2 is usually sufficient for clinical purposes. In Case 3, for example, the fact that the patient was hyperventilating and PaO2 was only 83 mm Hg indicates an elevated P(A-a)O2 and therefore a defect in gas exchange. The alveolar gas equation shows that with hyperventilation PaO2 should go up; PaO2 should be much higher than 83 mm Hg in a hyperventilating 27-year-old patient. Similarly, a patient breathing 40% oxygen whose PaO2 and PaCO2 are normal for room air (e.g., PaO2 90 mm Hg, PaCO2 40 mm Hg) has an elevated P(A-a)O2 and therefore a defect in gas exchange; with this FIO2, PAO2 should be over 200 mm Hg and PaO2 well over 100 mm Hg. These observations require nothing more than knowledge of the alveolar gas equation and simple mental calculation.
d) Since oxygen enters the pulmonary capillary blood by passive diffusion, it follows that in a steady state the alveolar PO2 must always be higher than the arterial PO2. This fact is useful to spot ‘garbage’ blood gas data, a not infrequent problem. For example, a PaO2 of 150 mm Hg in a patient breathing ‘room air’ at sea level (FIO2 = .21) must represent some kind of error, since at all conceivable PaCO2 values the P(A-a)O2 would have a negative value; even with extreme hyperventilation (PaCO2 10 mm Hg) the alveolar PO2 would be no higher than 140 mm Hg. A moment’s reflection will reveal several possible explanations for the apparently negative alveolar-arterial PO2 difference: the patient was in fact breathing supplemental oxygen during or just prior to the sample drawing; an air bubble in the arterial sample syringe; a quality control or reporting error from the lab; a transcription error – someone wrote down the wrong number; etc.
What about the oxygen values mentioned at the beginning of this section? A PaO2 of 28 mm Hg would be normal on the summit of Mt. Everest for a climber breathing ambient air. At the summit barometric pressure is 253 mm Hg, which provides a PIO2 of only 43 mm Hg24 (Table V).
TABLE V. Gas Pressures at Various Altitudes* LOCATION ALT. PB FIO2 PIO2 PaCo2 PAO2 PaO2 Sea Level 0 760 .21 150 40 102 95 Cleveland 500 747 .21 147 40 99 92 Denver 5280 640 .21 125 34 84 77 *Pikes’s Peak 14114 450 .21 85 30 62 55 *Mt. Everest 29028 253 .21 43 7.5 35 28 *All pressures in mm Hg; Pike’s Peak and Mt. Everest data from summits
ALT. = altitude in feet
PB = barometric pressure
FIO2 = fraction of inspired oxygen
PIO2 = pressure of inspired oxygen in the trachea
PaCO2 = arterial PCO2, assumed to = alveolar PCO2
PAO2 = alveolar PO2, PAO2 is calculated using an assumed R value of 0.8 except for the summit of Mt. Everest, where 0.85 is used 24
PaO2 = arterial PO2, assuming a P(A-a)O2 of 7 mm Hg at each altitude; each PaO2 value is normal for its respectove altitude
If the climber maintained PaCO2 at 40 mm Hg his PAO2 would be minus 5 mm Hg, a value wholly incompatible with life! Ability to oxygenate blood at this altitude without supplemental oxygen is made possible (in large part) by extreme hyperventilation. On one expedition to the summit, 10 minutes after supplemental oxygen was removed a climber’s end-tidal PCO2 (equivalent to PACO2) was measured at 7.5 mm Hg; assuming an R value of 0.85, the PAO2 was only 35 mm Hg.24 Based on a theoretical alveolar-arterial PO2 difference of 7 mm Hg, the climber’s PaO2 at the summit was estimated at 28 mm Hg – very low but ‘normal’ under the circumstances.24
A PaO2 of 55 mm Hg would likewise be normal at Pike’s Peak, Colorado, assuming a PaCO2 of 30 mm Hg from modest hyperventilation and a P(A-a)O2 of 7 mm Hg (Table V). On the other hand, a PaO2 of 95 mm Hg would represent a serious abnormality in anyone breathing 100% oxygen near sea level, as under these conditions PaO< sub> should be over 500 mm Hg. In summary, to properly interpret PaO2 one needs to have some appreciation of the alveolar PO< sub> , which requires knowing (at least approximately) the barometric pressure, FIO2 and PaCO2.
David Story finally confirmed something that was intuitively obvious and yet constantly denied by all clinicians I had spoken to–namely that PACO2 in the equation is merely a measure of minute ventilation and not a variable in the actual calculation (Anesthesiology 1996;84(4):1011)
4. Oxygen Content Equation Top
All physicians know that hemoglobin carries oxygen and that anemia can lead to severe hypoxemia. Making the necessary connection between PaO2 and O2 content requires knowledge of the oxygen content equation.
CaO2 = (SaO2 x Hb x 1.34) + .003(PaO2)
How much glucose is in the blood if the glucose level is 80 mm Hg? This question makes no sense, of course, because glucose is not a gas and therefore exerts no pressure in solution; any question regarding ‘how much’ is answered by determining its content, which in the case of glucose is usually reported as mg/dl blood. Oxygen is a gas and its molecules do exert a pressure but, like glucose, oxygen also has a finite content in the blood, in units of ml O2/dl blood. To remain viable tissues require a certain amount of oxygen per minute, a need met by a requisite oxygen content, not oxygen pressure. (Patients can and do live with very low PaO2 values, as long as their oxygen content and cardiac output are adequate.)
The oxygen carrying capacity of one gram of hemoglobin is 1.34 ml. With a hemoglobin content of 15 grams/dl blood and a normal hemoglobin oxygen saturation (SaO2) of 98%, arterial blood has a hemoglobin-bound oxygen content of 15 x .98 x 1.34 = 19.7 ml O2/dl blood. An additional small quantity of O2 is carried dissolved in plasma: .003 ml O2/dl plasma/mm Hg PaO2, or .3 ml O2/dl plasma when PaO2 is 100 mm Hg. Since normal CaO2 is 16-22 ml O2/dl blood, the amount contributed by dissolved (unbound) oxygen is very small, only about 1.4% to 1.9% of the total.
Given normal pulmonary gas exchange (i.e., a normal respiratory system), factors that lower oxygen content – such as anemia, carbon monoxide poisoning, methemoglobinemia, shifts of the oxygen dissociation curve – do not affect PaO2. PaO2 is a measurement of pressure exerted by uncombined oxygen molecules dissolved in plasma; once oxygen molecules chemically bind to hemoglobin they no longer exert any pressure.
PaO2 affects oxygen content by determining, along with other factors such as pH and temperature, the oxygen saturation of hemoglobin (SaO2). The familiar O2-dissociation curve can be plotted as SaO2 vs. PaO2 and as PaO2 vs. oxygen content (Figure 3). For the latter plot the hemoglobin concentration must be stipulated.
When hemoglobin content is adequate, patients can have a reduced PaO2 (defect in gas transfer) and still have sufficient oxygen content for the tissues (e.g., hemoglobin 15 grams%, PaO2 55 mm Hg, SaO2 88%, CaO2 17.8 ml O2/dl blood). Conversely, patients can have a normal PaO2 and be profoundly hypoxemic by virtue of a reduced CaO2. This paradox – normal PaO2 and hypoxemia – generally occurs one of two ways: 1) anemia, or 2) altered affinity of hemoglobin for binding oxygen.
A common misconception is that anemia affects PaO2 and/or SaO2; if the respiratory system is normal, anemia affects neither value. (In the presence of a right to left intrapulmonary shunt anemia can lower PaO2 by lowering the mixed venous oxygen content; when mixed venous blood shunted past the lungs mixes with oxygenated blood leaving the pulmonary capillaries, lowering the resulting PaO2.25 With a normal respiratory system mixed venous blood is fully oxygenated – as much as allowed by the alveolar PO2 – as it passes through the pulmonary capillaries.)
Obviously, however, the lower the hemoglobin content the lower the oxygen content. It is not unusual to see priority placed on improving a chronically hypoxemic patient’s low PaO2 when a blood transfusion would be far more beneficial.
Anemia can also confound the clinical suspicion of hypoxemia since anemic patients do not generally manifest cyanosis even when PaO2 is very low. Cyanosis requires a minimum quantity of de-oxygenated hemoglobin to be manifest – approximately 5 grams% in the capillaries.26,27 A patient whose hemoglobin content is 15 grams% would not generate this much reduced hemoglobin in the capillaries until the SaO2 reached 78% (PaO2 44 mm Hg); when hemoglobin is 9 grams% the threshold SaO2 for cyanosis is lowered to 65% (PaO2 34 mm Hg).27
Altered hemoglobin affinity may occur from shifts of the oxygen dissociation curve (e.g., acidosis, hyperthermia), from alteration of the oxidation state of iron in the hemoglobin (methemoglobinemia), or from carbon monoxide poisoning.
CASE 4. A 54-year-old man came to the emergency room (ER) complaining of headaches and shortness of breath. On room air his PaO2 was 89 mm Hg, PaCO2 38 mm Hg, pH 7.43; hematocrit was 44%. SaO2 was not directly measured but instead calculated at 98% for this PaO2, based on a standard oxygen dissociation curve. After some improvement he was scheduled for a brain CAT scan two days later, and discharged from the ER. He was brought back to the ER the next evening, unconscious. Ambulance attendants alerted the ER physician to a possible faulty heater in the patient’s house. This time carbon monoxide and SaO2 were measured along with routine arterial blood gases. The results: PaO2 79 mm Hg, PaCO2 31 mm Hg, pH 7.36, SaO2 53%, carboxyhemoglobin 46%.
This patient’s true SaO2 would have been much lower than 98% had it been measured on the first ER visit instead of just calculated. The physician missed hypoxemia as a cause of headache and dyspnea because of the ‘normal’ calculated SaO2.
Carbon monoxide by itself does not affect PaO2 but only SaO2 and O2 content. (Slight reduction in PaO2 on the patient’s second visit was attributed to some basilar atelectasis and resulting V-Q imbalance. The SaO2 and O2 content on the second visit are shown by an “X” in Figure 3.) Confusion about interpretation of oxygen saturation in the presence of excess CO is not unusual and even finds its way into peer-review literature.28
To know the oxygen content one needs to know the hemoglobin content and the SaO2; both should be measured as part of each arterial blood gas test. As shown above, a calculated SaO2 may be way off the mark and can be clinically misleading. This is true even without excess CO in the blood. One study of over 10000 arterial samples found wide variation in measured SaO2 for a given PaO2; for example, in the PaO2 range of 56-64 mm Hg the measured SaO2 ranged from 69.7 percent to 99.4 percent. 28
Finally, it should be noted that pulse oximeters are not reliable in the presence of dyshemoglobins – hemoglobins that cannot bind oxygen. The two major dyshemoglobins encountered in clinical practice are carboxyhemoglobin (COHb) and methemoglobin (Methb). Oximeters do not differentiate hemoglobin bound to carbon monoxide from hemoglobin bound to oxygen; the machines report the sum of both values as oxyhemoglobin.30-34
In contrast to blood co-oximeters, which utilize four wavelengths of light to separate out oxyhemoglobin from reduced hemoglobin, methemoglobin and carboxyhemoglobin, pulse oximeters utilize only two wavelengths of light 33-34. As a result, pulse oximeters measure COHb and part of any MetHb along with oxyhemoglobin, and combine the three into a single reading, the SpO2. (MetHb absorbs both wavelengths of light emitted by pulse oximeters, so that SpO2 is not affected as much by MetHb as for a comparable level of COHb).
Thus a patient with 80% oxyhemoglobin and 15% carboxyhemoglobin would show a pulse oximetry oxygen saturation (SpO2) of 95%, a value too high by 15%. For this reason pulse oximeters should be used cautiously (if at all) when there may be an elevated carbon monoxide level, for example in patients assessed in an emergency department. Note that excess carboxyhemoglobin is present in all cigarette and cigar smokers. A resting SpO2 should be interpreted cautiously in any outpatient who has smoked within 24 hours. The half- life of CO breathing ambient air is about 6 hours, so 24 hours after smoking cessation the CO level should be normal, i.e., less than 2.5%. If there is concern about the true SaO2, it should be measured on an arterial blood sample; alternatively, the percent COHb can be measured on a venous sample, and the value subtracted from the SpO2.
The spectrophotometric technique used by pulse oximeters also makes their oxygen saturation reading less reliable in the presence of excess methemoglobin (metHb). MetHb reduces the SpO2 linearly until a level of about 85%, at which point further increases in metHb do not cause further lowering of SpO2.35-37 A finding of unexpectedly low SpO2 (e.g., 91% in a patient with normal cardiorespiratory system who is receiving nasal oxygen) should make one think of excess methb; in such cases an arterial blood sample should be obtained for direct measurement of SaO2 and PaO2.
TRUE-FALSE QUIZ — based on “THE FOUR MOST IMPORTANT EQUATIONS IN CLINICAL PRACTICE”
Lawrence Martin, M.D., FACP, FCCP Top
Directions: This quiz is designed to test your understanding of information in the review paper, The Four Most Important Equations in Clinical Practice. For each of the following five numbered statements or questions, there are five lettered responses (a-e), each of which may be true or false.Circle the correct or true responses. Answers immediately follow the quiz.
1. Normal range for PaCO2 is 35-45 mm Hg. A change in PaCO2 from normal
to 28 mm Hg means the subject
- a) is hyperventilating.
- b) has excess alveolar ventilation for the amount of CO2 production.
- c) must have hypoxia, anxiety, and/or metabolic acidosis.
- d) must be breathing faster than normal.
- e) must have acute respiratory alkalosis.
2. The arterial PO2 is predicted to be reduced to some extent from
- a) anemia.
- b) ventilation-perfusion (V-Q) imbalance with an increase in the number of low V- Q units.
- c) increased PCO2, while the subject is breathing room air.
- d) carbon monoxide poisoning.
- e) altitude.
3. To obtain a reasonable idea of the acid-base state of a patient’s blood,
you would need to know the
- a) pH and PaCO2.
- b) pH and PaO2.
- c) PaCO2 and PaO2.
- d) PaCO2 and HCO3-.
- e) pH and SaO2 (%saturation of hemoglobin with oxygen).
4. Arterial blood gas data (pH, PaCO2, PaO2, SaO2) are related in some simple
but important ways. Which of the following are valid relationships?
- a) Alveolar PO2 is related to PaCO2 by the alveolar gas equation: as PaCO2 goes up, alveolar PO2 goes down.
- b) PaO2 is inversely related to blood pH: as pH goes up, PaO2 goes down.
- c) If PaCO2 increases while HCO3- remains unchanged, pH always goes down.
- d) PaO2 is related to SaO2 on a linear scale (i.e., a straight-line relationship).
- e) The SaO2 is related to hemoglobin-bound arterial oxygen content on a linear scale (i.e., a straight-line relationship).
5. There are some “truisms” in terminology and physiology for proper blood gas interpretation. They include which of the following?
- a) “Hyperventilation” and “hypoventilation” are clinical terms, and are not diagnosed by arterial blood gases.
- b) The alveolar-arterial PO2 difference increases with age and with increase in the fraction of inspired oxygen.
- c) The arterial PO2 cannot go above 100 mm Hg while breathing room air at sea level.
- d) A continuously negative alveolar-arterial PO2 difference is incompatible with life.
- e) If arterial pH is normal, the patient cannot have a clinically significant acid-base disorder.
A N S W E R S
1. a and b are true. The patient may have hyperventilation from many causes (including voluntary hyperventilation). The subject may be breathing deeper than normal, rather than faster. And the subject may be hyperventilating to compensate for metabolic acidosis, which would not be a respiratory alkalosis.
2. b, c and e are true; other responses are false. Anemia and carbon monoxide poisoning do not affect PaO2 (except when there is a ventilation-perfusion imbalance and some right to left shunting).
3. a and d are true. You need to know two of the three Henderson-Hasselbalch equation variables to assess acid-base status.
4. a, c and e are true. PaO2 and pH are not related in any formal way. The relationship of PaO2 to SaO2 is sigmoid-shaped, not straight line.
5. b and d are true. Hyperventilation and hypoventilation are specifically not defined by clinical or bedside criteria, but by changes in PaCO2. The PaO2 can easily go above 100 mm Hg with hyperventilation and normal lungs. Arterial pH can be normal with two or more acid-base disorders occurring at the same time.
R E F E R E N C E S Top
1. Martin L. Pulmonary Physiology in Clinical Practice.C.V. Mosby Co., St. Louis, 1987.
2. Wasserman K, Whipp BJ. Exercise physiology in health and disease: State of the Art. Amer Rev Resp Dis 1975;112:219-49.
3. Flenly DC. Another non-logarithmic acid-base diagram? The Lancet 1971;1:691-65.
4. McCurdy DK. Mixed metabolic and respiratory acid-base disturbances: diagnosis and treatment. Chest 1972;62:35S-44S.
<>6. Arbus GS. An in-vivo acid-base nomogram for clinical use. CMA Jour 1973;291-2.
<>7. Fulop M, Fulop M. Acid-base diagrams. Maths, myths and measurements. The Lancet 1974;2:637-9.
<>9. Winters RW. Terminology of acid-base disorders. Ann Intern Med 1965;63:873-84.
<>15. Javaheri S. Compensatory hypoventilation in metabolic alkalosis. Chest 1982;81:296-301.
<>25. Dantzker, DR. Cardiopulmonary Critical Care. Grune & Stratton, Orlando, 1986; page 39.
<>26. Lundsgaard C, Van Slyke DD. Cyanosis. Medicine 1923;2:1-76.
<>28. Hampson NB. Arterial oxygenation in carbon monoxide poisoning (Letter). Chest 1990;98:1538-9.
<>34. Principles of Pulse Oximetry. Clinical Monograph. Nellcor Corp., Pleasanton, CA, 1991.
Other Equations
Desired MV = (Current PaCO2 x Current MV) / Desired PaCO2